Arrhythmias
Fetal cardiac rhythm disturbances may be broadly categorized as tachyarrhythmias, heart rates > 180 beats per minute (bpm); bradyarrhythmia, heart rate < 110 bpm; and ectopy, typically premature atrial contractions. Fetal M-mode sonography (p. 322) should be performed to determine the atrial and ventricular rate to clarify the relationship between atrial and ventricular beats, thereby diagnosing the rhythm disturbance type.
Premature Atrial Contractions
This is by far the most common arrhythmia. Premature atrial contractions are identified in 1 to 2 percent of pregnancies and are generally a benign finding (Hahurij, 2011; Strasburger, 2010). They represent immaturity of the cardiac conduction system and typically resolve either with advancing gestation or in the neonatal period. Although they may be conducted, they are more commonly blocked, and with handheld Doppler or fetoscope, they sound like a dropped beat. Premature atrial contractions are not associated with major structural cardiac abnormalities, but they sometimes occur with an atrial septal aneurysm. As shown in Figure 10-24 (p. 212), M-mode evaluation demonstrates that the dropped beat is a compensatory pause following the premature atrial contraction. They may occur as frequently as every other beat, known as blocked atrial bigeminy. This results in an auscultated fetal ventricular rate as low as 60 to 80 beats per minute. Unlike other causes of bradycardia, this carries a benign prognosis and does not require treatment (Strasburger, 2010).
Approximately 2 percent of fetuses with premature atrial contractions are later identified to develop supraventricular tachycardia (SVT) and require urgent treatment to prevent development of hydrops (Copel, 2000; Srinivasan, 2008). Given the importance of identifying such tachycardia, the fetus with premature atrial contractions is often monitored with heart rate assessment every 1 to 2 weeks until ectopy resolution.
Tachyarrhythmias
The two most common are supraventricular tachycardia and atrial flutter. SVT is characterized by an abrupt increase in the fetal heart rate to 180 to 300 bpm with 1:1 atrioventricular concordance. The typical range is 200 to 240 bpm. SVT may develop secondary to an ectopic focus or to an accessory atrioventricular pathway leading to a reentrant tachycardia. Atrial flutter is characterized by a much higher atrial rate—300 to 500 bpm. There are varying degrees of atrioventricular block, such that the ventricular rate may range from below normal to approximately 250 bpm (Fig. 16-1). In contrast, fetal sinus tachycardia typically presents with a gradual heart rate rise to a rate slightly above normal. There is often a readily discernible cause such as maternal fever or hyperthyroidism, or rarely, fetal anemia or infection.
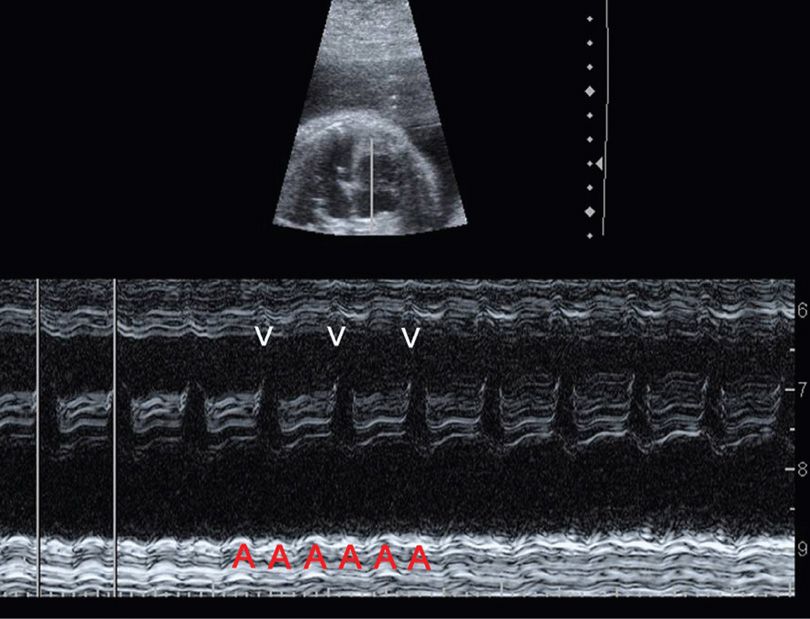
FIGURE 16-1 Atrial flutter. In this M-mode image at 28 weeks, calipers mark the ventricular rate, which is approximately 225 bpm. There are two atrial beats (A) for each ventricular beat (V), such that the atrial rate is approximately 450 bpm with 2:1 atrioventricular block.
If a tachyarrhythmia is identified, it is important to determine whether it is sustained—defined as present for at least 50 percent of the time. It may be necessary to monitor the fetal heart rate for 12 to 24 hours upon initial detection, and then periodically to reassess (Srinivasan, 2008). Nonsustained or intermittent tachyarrhythmias generally do not require treatment, provided that fetal surveillance is reassuring.
Sustained fetal tachyarrhythmia with ventricular rates exceeding 200 bpm impairs ventricular filling to such a degree that the risk of hydrops is significant. With atrial flutter, lack of coordinated atrioventricular contractions may further compound this risk. Maternal administration of antiarrhythmic agents that cross the placenta may convert the rhythm to normal or lower the baseline heart rate to forestall heart failure. Therapy may require dosages at the upper end of the therapeutic adult range. A maternal electrocardiogram should be performed before and during therapy. If the fetus has become hydropic, the drug may need to be administered directly via the umbilical vein (Mangione, 1999; Simpson, 2006).
Various antiarrhythmic medications have been used, most commonly digoxin, sotalol (Betapace), flecainide (Tambocor), and procainamide (Pronestyl). Their selection depends on the type of tachyarrhythmia as well as provider familiarity and experience with the drug. Digoxin is usually the first-line agent. Amiodarone (Cordarone) has been associated with fetal and neonatal hypothyroidism, which may be severe (Niinikoski, 2007; Simpson, 2006).
In a review of 485 cases of fetal tachyarrhythmia, hydrops was reported in approximately 40 percent of those with either SVT or atrial flutter (Krapp, 2003). Digoxin was the first-line agent selected in two thirds with either tachyarrhythmia. Treatment was more effective in nonhydropic fetuses than in those in whom hydrops had already developed. With therapy, the neonatal survival rate with either arrhythmia exceeded 90 percent (Krapp, 2003).
Bradyarrhythmia
The most common etiology of pronounced fetal bradycardia is congenital heart block. Approximately 50 percent of cases occur in the setting of a structural cardiac abnormality involving the conduction system. These include heterotaxy, in particular left-atrial isomerism; endocardial cushion defect; and corrected transposition of the great vessels (Srinivasan, 2008). The prognosis of heart block secondary to a structural cardiac anomaly is extremely poor, and fetal loss rates exceed 80 percent (Glatz, 2008; Strasburger, 2010). In a structurally normal heart, 85 percent of cases of atrioventricular block develop secondary to transplacental passage of maternal anti-SSA/Ro or anti-SSB/La antibodies (Buyon, 2009). Many of these women have, or subsequently develop, systemic lupus erythematosus or another connective-tissue disease (Chap. 59, p. 1172). The risk of third-degree heart block with these antibodies is only 2 to 5 percent, but it may be up to 20 percent if a prior infant has been affected. Immune-mediated congenital heart block confers a mortality rate of 20 to 30 percent, requires permanent pacing in two thirds of surviving children, and also poses a risk for cardiomyopathy (Buyon, 2009). If associated with effusions, bradyarrhythmias, or endocardial fibroelastosis, neonatal status may progressively worsen after birth (Cuneo, 2007).
Research efforts have focused on maternal corticosteroid therapy to reverse fetal heart block or forestall it. Friedman and colleagues (2008, 2009) conducted a prospective multicenter trial of pregnancies with anti-SSA/Ro antibodies—the PR Interval and Dexamethasone (PRIDE) study. Weekly sonography was used to monitor for fetal heart block. If identified, maternal treatment was dexamethasone 4 mg orally daily. There were several important findings. First-degree block was rare and did not generally precede to more advanced block; progression from second- to third-degree block was not prevented with maternal dexamethasone therapy; and third-degree atrioventricular block was irreversible (Friedman, 2008, 2009). In rare cases, there was a potential benefit in reversing first-degree atrioventricular block. However, the authors cautioned that there was a need to weigh this potential benefit against the risks of chronic antepartum corticosteroid treatment, including impaired fetal growth (Friedman, 2009).
Maternal terbutaline has also been administered to increase the fetal heart rate in cases with sustained bradycardia of any cause in which the fetal heart rate is ≤ 55 bpm. Cuneo and associates (2007) reported reversal of hydrops with this therapy in a few cases. However, outcomes in those with structural abnormalities remained poor.
 Congenital Adrenal Hyperplasia
Congenital Adrenal Hyperplasia
Several autosomal recessive enzyme deficiencies cause impaired fetal synthesis of cortisol from cholesterol by the adrenal cortex, resulting in congenital adrenal hyperplasia (CAH). This is the most common etiology of androgen excess in females with pseudohermaphroditism (Chap. 7, p. 148). Lack of cortisol stimulates adrenocorticotrophic hormone (ACTH) secretion by the anterior pituitary, and the resulting androstenedione and testosterone overproduction leads to virilization of female fetuses. Sequelae may include formation of labioscrotal folds, development of a urogenital sinus, or even creation of a penile urethra and scrotal sac (Fig. 7-18, p. 149).
More than 90 percent of CAH cases are caused by 21-hydroxylase deficiency, which is found in classic and nonclassic forms. The incidence of classic CAH is approximately 1:15,000 overall and is higher in selected populations. For example, it develops in approximately 1:300 Yupik Eskimos (Nimkarn, 2010). Among those with classic CAH, 75 percent are at risk for salt-wasting adrenal crises and require postnatal treatment with mineralocorticoids and glucocorticoids. The remaining 25 percent with classic CAH have the simple virilizing type and require glucocorticoid supplementation. Without prompt recognition and treatment, infants with the salt-wasting form can develop hyponatremia, dehydration, hypotension, and cardiovascular collapse. As discussed in Chapter 32 (p. 631), all states mandate newborn screening for CAH.
The efficacy of maternal dexamethasone treatment to suppress fetal androgen overproduction and either obviate or ameliorate virilization of female fetuses has been recognized for nearly 30 years (David, 1984; New, 2012). Prenatal corticosteroid therapy is considered successful in 80 to 85 percent of cases (Miller, 2013; Speiser, 2010). The alternative is consideration of postnatal genitoplasty, a complex and somewhat controversial surgical procedure, which may involve vaginoplasty, clitoroplasty, and labioplasty (Braga, 2009).
If treatment is elected, the typical regimen is oral dexamethasone given to the mother at a dosage of 20 μg/kg/d—up to 1.5 mg per day, divided in three doses. The critical period for external genitalia development is 7 to 12 weeks’ gestation. To prevent virilization, treatment should be initiated by 9 weeks, before it is known whether the fetus is at risk. Because this is an autosomal recessive condition, affected females comprise only 1 in 8 at-risk conceptions.
Typically, carrier parents are identified after the birth of an affected child. Molecular genetic testing is clinically available for common mutations and deletions of the CYP21A2 gene, which encodes the 21-hydroxylase enzyme. It is informative in 80 to 98 percent of cases, and for the remainder, gene sequencing may detect rarer alleles (Nimkarn, 2010). Women who elect treatment do so with a plan to undergo prenatal diagnosis and stop therapy if the fetus is male or an unaffected female. Prenatal diagnosis with molecular genetic testing may be performed on chorionic villi—at 10 to 12 weeks’ gestation—or on amniocytes after 15 weeks. Ideally, the goal is to limit dexamethasone exposure in males and in unaffected females.
Recently, maternal treatment with dexamethasone has become a topic of significant controversy. The Endocrine Society has recommended that treatment be given only in the context of research protocols (Miller, 2013; Speiser, 2010). If therapy is initiated shortly before 9 weeks, the dosage of dexamethasone used is not considered to have significant teratogenic potential because organogenesis of major organs has already taken place (McCullough, 2010). There are ongoing concerns, however, about the potential effects of either excess endogenous androgens or excess exogenous dexamethasone on the developing brain. Although maternal dexamethasone has been used for many years to prevent virilization of female fetuses with CAH, long-term safety data are relatively limited.
Development of cell-free fetal DNA testing of maternal serum has potential to replace invasive tests such as chorionic villus sampling and amniocentesis for CAH (Chap. 13, p. 279). Determination of fetal gender using cell-free fetal DNA is reported to have at least 95-percent sensitivity when performed at or beyond 7 weeks’ gestation (Devaney, 2011). Although not yet clinically available in 2013, DNA analysis of the CYP21A2 gene has also been reported using cell-free fetal DNA testing.
 Congenital Cystic Adenomatoid Malformation
Congenital Cystic Adenomatoid Malformation
Also called congenital pulmonary airway malformation (CPAM), this lung mass is a hamartomatous overgrowth of terminal bronchioles. Sonographically, congenital cystic adenomatoid malformation (CCAM) is a well-circumscribed mass that may appear solid and echogenic or may have one or multiple variably sized cysts (Fig. 10-18, p. 207). Lesions with cysts 5 mm or larger are generally termed macrocystic, whereas microcystic lesions have smaller cysts or appear solid (Adzick, 1985). Therapy for macrocystic CCAM is discussed on page 330.
Infrequently, a microcystic CCAM may demonstrate rapid growth between 18 and 26 weeks’ gestation. The mass may become so large that it causes mediastinal shift, compromised cardiac output and venous return, and resultant hydrops (Cavoretto, 2008). A CCAM-volume ratio (CVR) has been used to quantify size and risk for hydrops in these severe cases (Crombleholme, 2002). The CVR is an estimate of the CCAM volume—length × width × height × 0.52—divided by the head circumference. In the absence of a dominant cyst, a CVR exceeding 1.6 has been associated with a hydrops risk of approximately 75 percent, whereas the risk is < 5 percent with a smaller CVR.
If the CVR exceeds 1.4 or 1.6 or if signs of hydrops develop, corticosteroid treatment has been advocated to improve outcome. Regimens include dexamethasone—6.25 mg every 12 hours for four doses, or betamethasone—12.5 mg intramuscularly every 24 hours for two doses. In some reports, hydrops has resolved in nearly 80 percent of cases, and approximately 85 percent of these treated cases survived (Curran, 2010; Loh, 2012; Peranteau, 2007). However, others found that high-risk CCAMs displayed a variable response to corticosteroid treatment, and perinatal mortality rates were > 40 percent (Morris, 2009c).
 Thyroid Disease
Thyroid Disease
Identification of fetal thyroid disease is rare and usually prompted by sonographic detection of a fetal goiter. There are several possibilities to consider. If a woman has previously been treated for Graves disease with either thyroid ablation or surgical resection, she may continue to produce IgG thyroid-stimulating immunoglobulins. These cross the placenta to cause fetal thyrotoxicosis. In a woman receiving medication for Graves disease, transplacental passage of propylthiouracil or methimazole usually prevents this, as discussed in Chapter 58 (p. 1149). Occasionally, maternal treatment—or overtreatment—for thyrotoxicosis may cause fetal hypothyroidism (Bliddal, 2011a). Other potential causes of fetal hypothyroidism resulting in goiter include transplacental passage of thyroid peroxidase antibodies and fetal thyroid dyshormonogenesis (Agrawal, 2002).
If a goiter is identified, it is important to determine whether the fetus is hyper- or hypothyroid. Thyroid hormone levels may be measured in amnionic fluid, but fetal blood sampling, discussed in Chapter 14 (p. 300), is preferred for guiding treatment (Abuhamad, 1995; Ribault, 2009). A primary purpose of therapy—in addition to correcting the physiological abnormality—is to decrease goiter size. The goiter may compress the trachea and esophagus to such a degree that the fetus may develop severe hydramnios and the neonate may develop airway compromise. Hyperextension of the fetal neck by a goiter may result in labor dystocia.
Fetal Thyrotoxicosis
Untreated fetal thyrotoxicosis may present with goiter, tachycardia, growth restriction, hydramnios, accelerated bone maturation, and even heart failure and hydrops (Huel, 2009; Peleg, 2002). The cause is usually Graves disease with transplacental passage of thyroid-stimulating immunoglobulins. Most recommend fetal blood sampling to confirm the diagnosis (Duncombe, 2001; Heckel, 1997; Srisupundit, 2008). Confirmed fetal thyrotoxicosis is followed by maternal antithyroid treatment, and if the mother develops hypothyroidism, she is given supplemental levothyroxine (Hui, 2011).
Fetal Hypothyroidism
Goitrous hypothyroidism may lead to hydramnios, neck hyperextension, and delayed bone maturation. If the mother is receiving antithyroid medication, discontinuation is generally recommended, along with intraamnionic levothyroxine injection (Bliddal, 2011a; Ribault, 2009). There have been numerous case reports of intraamnionic levothyroxine treatment. However, optimal dosage and frequency have not been established, and reported dosages range from 50 to 800 mg every 1 to 4 weeks (Abuhamad, 1995; Bliddal, 2011b; Ribault, 2009).
 Fetal Stem-Cell Transplantation
Fetal Stem-Cell Transplantation
In theory, stem-cell transplantation could be used to treat various hematological, metabolic, and immunological diseases. It could also serve as a delivery vehicle for gene transfer to treat other genetic conditions. The fetal period is ideal for this because in the first and early second trimesters, the fetus lacks an adaptive immune response to foreign antigens—described as preimmune (Tiblad, 2008). Also, pretreatment chemotherapy or radiation is not necessary before transplantation, and graft-versus-host disease is less likely.
Fetal stem-cell transplantation has been most successful in the treatment of immunodeficiency syndromes. Engraftment has been achieved in fetuses with severe combined immunodeficiency and bare lymphocyte syndrome (Tiblad, 2008). Treatment of red blood cell and metabolic disorders, however, has not been successful (Mummery, 2011). Stem-cell transplantation has been attempted to treat hemoglobinopathies without success. Some children with α- and β-thalassemia have remained transfusion dependent despite achieving engraftment (Tiblad, 2008; Westgren, 1996). One fetus with osteogenesis imperfecta, type II, was transplanted with fetal mesenchymal stem cells and developed both engraftment and chimerism (Le Blanc, 2005; Mummery, 2011). Although long-term outcomes of such cases remain uncertain, the technology holds great promise.
SURGICAL THERAPY
Also called maternal-fetal surgery, these procedures are offered for selected congenital abnormalities in which the likelihood of fetal deterioration is so great that delaying treatment until after delivery would risk fetal death or substantially greater postnatal morbidity (Walsh, 2011). Open fetal surgery is a highly specialized, multidisciplinary intervention performed at relatively few centers in the United States and for only a few fetal conditions. It was pioneered more than three decades ago by Harrison and coworkers (1982) at the University of California, San Francisco. Criteria for consideration of fetal surgery are listed in Table 16-1. In many cases, data regarding the safety and efficacy of these procedures are lacking. The Agency for Healthcare Research and Quality has stressed that when considering fetal surgery, the overriding concern must be maternal and fetal safety. Accomplishing the fetal goals of the procedure are secondary (Walsh, 2011).
TABLE 16-1. Guiding Principles for Fetal Surgical Procedures
Accurate prenatal diagnosis for the defect is available, with staging if applicable
The defect appears isolated, with no evidence of other abnormality or underlying genetic syndrome that would significantly worsen survival or quality of life
The defect results in a high likelihood of death or irreversible organ destruction, and postnatal therapy is inadequate
The procedure is technically feasible, and a multidisciplinary team is in agreement regarding the treatment plan
Maternal risks from the procedure are well documented and considered acceptable
There is comprehensive parental counseling
It is recommended that there is an animal model for the defect and procedure
Some abnormalities amenable to fetal surgical treatment, antepartum or intrapartum, are shown in Table 16-2. Information regarding the procedures, their indications, and complications is provided to assist patient evaluation and counseling. It is beyond the scope of this text to provide technical information necessary to perform these procedures or address individual circumstances in which their benefits may outweigh potential risks.
TABLE 16-2. Selected Fetal Abnormalities Amenable to Fetal Surgery
Open Fetal Surgery
Myelomeningocele
Congenital cystic adenomatoid malformation (CCAM)
Extralobar pulmonary sequestration
Sacrococcygeal teratoma
Fetoscopic Surgery
Twin-twin transfusion: laser of placental anastomoses
Diaphragmatic hernia: fetal endoscopic tracheal occlusion (FETO)
Posterior urethral valves: cystoscopic laser
Congenital high airway obstruction: vocal cord laser
Amnionic band release
Percutaneous Procedures
Shunt therapy
Posterior-urethral valves/bladder outlet obstruction
Pleural effusion: chylothorax or sequestration
Dominant cyst in CCAM
Radiofrequency ablation
Twin-reversed arterial perfusion (TRAP) sequence
Monochorionic twins with severe anomaly(ies) of 1 twin
Chorioangioma
Fetal intracardiac catheter procedures
Aortic or pulmonic valvuloplasty for stenosis
Atrial septostomy for hypoplastic left heart with restrictive atrial septum
Ex-Utero Intrapartum Treatment (EXIT) Procedures
Congenital diaphragmatic hernia after FETO
Congenital high airway obstruction sequence (CHAOS)
Severe micrognathia
Tumors involving neck or airway
EXIT-to-resection: resection of fetal thoracic or mediastinal mass
EXIT-to-extracorporeal membrane oxygenation (ECMO): congenital diaphragmatic hernia
 Open Fetal Surgery
Open Fetal Surgery
These procedures require a highly-skilled multidisciplinary team and extensive preoperative counseling. The mother must undergo general endotracheal anesthesia to suppress both uterine contractions and fetal responses. Using sonographic guidance to avoid the placental edge, a hysterotomy incision is made with a stapling device that seals the edges for hemostasis. To replace amnionic fluid losses, warmed fluid is continuously infused into the uterus thorough a rapid infusion device. The fetus is gently manipulated to permit pulse oximetry monitoring and venous access, in case fluids or blood are emergently needed. The surgical procedure is then performed. After completion, the hysterotomy is closed and tocolysis begun. Tocolysis typically includes intravenous magnesium sulfate for 24 hours, oral indomethacin for 48 hours, and at some centers, oral nifedipine until delivery (Wu, 2009). Prophylactic antibiotics are also administered and generally continued for 24 hours following the procedure. Cesarean delivery will be needed later in gestation and for all future deliveries.
Risks
Morbidities associated with fetal surgery have been well characterized. In a review of 87 open procedures from the University of California, San Francisco, Golombeck and colleagues (2006) reported the following morbidities: pulmonary edema—28 percent, placental abruption—9 percent, blood transfusion—13 percent, premature rupture of membranes—52 percent, and preterm delivery—33 percent. Wilson and associates (2010) from Children’s Hospital of Philadelphia reviewed subsequent pregnancy outcomes following open fetal surgery and reported that 14 percent experienced uterine rupture and 14 percent had uterine dehiscence. Morbidities identified in the recent Management of Myelomeningocele Study—MOMS—are shown in Table 16-3 (Adzick, 2011). Other potential risks include maternal sepsis and fetal death during or following the procedure.
TABLE 16-3. Benefits and Risks of Fetal Myelomeningocele Surgery versus Postnatal Repair
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


