(a) Normal face and lips, (b) unilateral and (c) bilateral cleft lips.
Diagnosis
A coronal section through the face reveals the lips, chin prominence and nostrils, often in the same plane. A sagittal profile view may show a ‘proboscis’-like appearance of the upper lip. This appearance is caused by the lack of attachment of the unpaired frontonasal process to the paired bilateral maxillary processes. In the case of cleft palate, color Doppler examination during fetal swallowing or breathing movement may reveal mixing of fluid in the nasal and buccal cavity.
The advent of three-dimensional (3D) scanning has allowed a better representation of the defect and may be helpful for the maxillofacial surgeons in advising parents on prognosis and the type of surgery required.
Cause
The majority of cases are multifactorial in nature. However, there is a strong association with chromosomal and syndromic malformations. A variety of chromosomal abnormalities have been reported in 3–4% of cases of cleft lip and cleft palate, and 1–2% of cases of cleft palate alone. Hence, prenatal karyotyping should be considered in all cases following the diagnosis of orofacial cleft. However, even isolated cleft lip and palate does show a familial tendency, although this is likely to be multifactorial rather than a single gene defect. Some evidence is coming to light that folic acid deficiency may play a role in cleft lip and palate[2–4].
Smoking and alcohol consumption have been implicated in the incidence of cleft lip and palate, with smoking increasing the risk by twofold. The antiepileptic medications phenytoin and carbamazepine are associated with cleft lip and palate. Other drugs include methotrexate and retinoids, such as vitamin A. Rarely, amniotic band syndrome can cause bizarre patterns of facial clefts.
Management
Once a diagnosis of cleft lip or palate is made, a search should be performed for associated abnormalities, particularly congenital heart disease; 5–12% of cases with cleft lip and palate have an associated congenital heart defect[5]. Karyotyping should be offered, particularly if the cleft lip or palate is midline or bilateral or there are associated abnormalities, as these have a higher chance of a chromosomal issue.
A further ultrasound scan can be performed in the third trimester to detect further abnormalities that may become more obvious in later pregnancy or fetal growth restriction. Mild polyhydramnios may be noted but is usually of no clinical consequence.
The parents should be referred to a specialist cleft team and offered support through a specialist organization. A multidisciplinary approach to neonatal management with neonatologists and specialist cleft surgeons should be discussed with the parents.
Outcome
If the cleft lip or palate is isolated, there is no need to deliver at a tertiary level center unless there are associated abnormalities. The main issue after delivery will be the establishment of feeding, and there are multiple specialized devices available, such as bottles with longer teats.
The majority of cleft lip and palates have a good outcome if isolated, and modern cosmetic surgery is associated with a good result. All cleft lips and palates will need some form of surgical repair. Three surgical procedures are usually required. The first operation is usually performed at around 10 weeks to close the external lip and facial soft tissues. The second operation is usually performed at 6–12 months and involves the closure of deeper palatal and alveolar defects to allow speech and language development. A third procedure is performed at 6–10 years to correct alveolar bony defects. A final procedure may be required in the late teen period to revise the jaw, lip or nose.
Recurrence
Recurrence depends on the underlying cause, but if isolated, varies from 2–5% if one first-degree relative is affected and up to 10% if a parent and sibling are affected (Table 7.1). Folic acid prenatally may reduce this risk[6].
| Relationship to index case | Recurrence risk (%) |
|---|---|
| Sibling unilateral cleft lip | 2–3 |
| Sibling unilateral cleft lip and palate | 4 |
| Sibling bilateral cleft lip and palate | 5–6 |
| Two affected siblings | 10 |
| Affected sibling and parent | 10 |
| Affected parent | 4 |
Micrognathia (small jaw)
Micrognathia is characterized by a small mandible and a receding chin. It has an incidence of 1 in 5,000 to 1 in 10,000 deliveries. There are a large number of syndromes and chromosomal abnormalities that can be associated with micrognathia.
Diagnosis
Micrognathia can be difficult to assess as it is largely subjective. It can only truly be assessed in the profile view. It is worth noting that it is very easy to create false micrognathia if an oblique profile view is obtained. Again, as in facial clefts, the use of 3D scanning may help to visualize the facial profile and may be used in counseling the parents on outcome.
Micrognathia is associated with a wide range of chromosomal abnormalities, in particular, trisomies 18 and 13. In fact, about 60% will have some form of chromosomal issue. There are also a wide range of syndromic abnormalites associated with micrognathia, which include Treacher–Collins syndrome, an autosomal dominant condition with micrognathia and hearing loss, Pierre Robin syndrome, which has an associated cleft palate, Stickler syndrome, which is a collagen gene disorder, as well as a strong association with cardiac abnormalities due to its association with other syndromes, such as DiGeorge or 22q11 deletion syndrome[8].
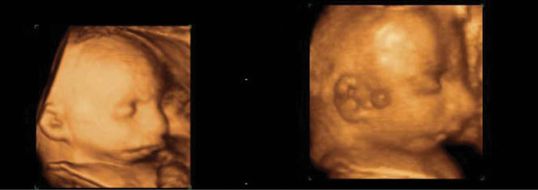
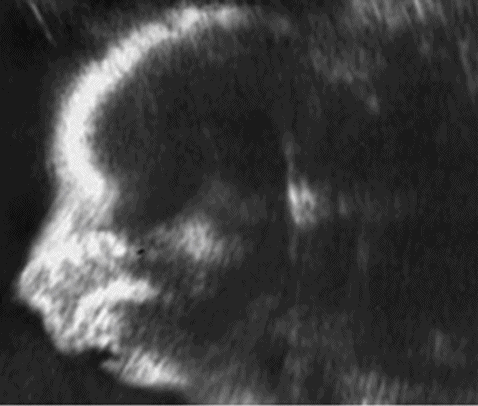
(a–c) Three- and two-dimensional ultrasound images of micrognathia in a fetus with Pierre Robin syndrome.
Management
In cases of micrognathia, full karyotyping is essential due to the wide range of chromosomal associations. A careful secondary survey looking for other abnormalities should be performed, and a fetal echocardiogram is also important, with the aim to establish an underlying genetic problem or syndrome. If other abnormalities are detected, a geneticist should be involved to determine the underlying diagnosis.
The main antenatal issue is that of polyhydramnios, which may be severe due to the inability of the jaw to open and swallow fluid, causing preterm delivery, and may require amniodrainage. Further scanning should be performed at regular intervals throughout the pregnancy to detect polyhydramnios, as well as to search for other structural abnormalities that may become apparent later in pregnancy, and to diagnose fetal growth restriction due to the underlying chromosomal or syndromic issue[9].
When diagnosed prenatally, a discussion with neonatalogists and pediatric ear, nose and throat surgeons is essential. Delivery should occur at a center with the ability to provide expertise in dealing with difficult intubations and with the ability to perform emergency tracheostomy. It may be that the fetus needs to be delivered electively by cesarean section so that the appropriate pediatric team are available to deal with a difficult intubation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


