Condition
Explanation
There is no medically equivalent histocompatible adult relative who is willing and able to donate
An adult will be better able to understand the rationale for donation, the procedural details of donation, and its risks and benefits, more closely approximating ideal informed consent
There is a strong personal and emotionally positive relationship between the donor and recipient
This increases the likelihood that the donor will experience psychological benefits from donation
There is some likelihood that the recipient will benefit from transplantation
If the chance of successful transplantation are below some minimum threshold (not defined), the donor should not be exposed to the medical and psychological risks of donationa
The clinical, emotional, and psychosocial risks to the donor are minimized and are reasonable in relation to the benefits expected to accrue to the donor and to the recipient
This ensures that parents consider the benefits and burdens of sibling donation from the independent perspectives of the donor and recipient
Parental permission and, where appropriate, child assent have been obtained
As with all informed consent, this respects patient autonomy
We must acknowledge the conflicts of interest unique to sibling HSC donationa:
Parents face the tension of exposing their healthy child to modest risks, with the hope of saving the life of their child who is ill
Transplant teams primary responsibility is to the recipient; however, the same providers may participate in the evaluation, consent, and treatment of the donor
Ethical Issues in Human Subjects Research
Progress in the field of pediatric oncology is often touted as the archetype of the improved health outcomes attainable through coordinated research efforts of clinicians and scientists around the world (O’Leary et al. 2008). Indeed, ever since Sidney Farber first induced remission in a child with acute lymphoblastic leukemia (ALL) using aminopterin (Farber and Diamond 1948), advances in cancer care now give us greater than 85 % chance of cure for pediatric ALL (Howlader et al. 2014). Despite this success and the promise research offers, the scientific community faces numerous ethical dilemmas in their efforts to cure pediatric cancer.
Historical Background
For more than a century, scholars have advocated for the need to protect children as a vulnerable population who could fall victim to unethical research (Bercovici 1921; Grodin and Glantz 1994). Yet it was not until after World War II, when Nazi experimentation on prisoners was revealed, that the scientific community began to codify ethical standards for medical research, in what is known as the Nuremberg Code (International Military 1949). Without explicitly mentioning children, its emphasis on informed consent provided some guidance until subsequent codes did. In 1964 the World Medical Association’s Declaration of Helsinki outlined additional ethical research principles beyond informed consent, such as the concept that the “interests of the subject must always prevail over the interest of science and society” (World Medical Association 1997).
In 1974, as details of the controversial Syphilis Study at Tuskegee emerged, the US Congress created the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. The Commission published the Belmont Report, outlining three basic ethical principles, respect for persons, beneficence, and justice, which form the framework for ethical discourse in biomedical research and practice (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research 1978). Respect for persons embodies the concept of autonomy, requiring that one’s preferences be respected. A corollary is that “persons with diminished autonomy [e.g., children] are entitled to protection.” Beneficence is defined as an obligation to carry out actions that “(1) do not harm and (2) maximize possible benefits and minimize possible harms.” Lastly, justice encompasses the concept that risks and benefits ought to be distributed fairly and equally and that no individual be denied benefits to which they are entitled or forced to shoulder undue burden (Beauchamp and Childress 2013). The report outlined the processes of informed consent, risk/benefit assessment, and fair selection of subjects as procedural safeguards to these principles.
The terms “research” and “human subject” are best defined by the Code of Federal Regulations regarding human subjects research. Research is “a systematic investigation designed to develop or contribute to generalizable knowledge,” and a human subject is “a living individual about whom an investigator conducting research obtains: (1) data through intervention or interaction with the individual, or (2) identifiable private information” (Office for Protection from Research Risks 1983). The regulations include the requirement that an Institutional Review Board (IRB) reviews all research activities. However, members of the scientific community itself have worked to articulate a comprehensive rubric to determine whether research protocols are ethical (Table 17.2).
Table 17.2
Requirements for determining whether human subjects research is ethical
Principle | Explanation |
|---|---|
Social or scientific value | Research evaluates a treatment, intervention, or theory with the potential to improve health, well-being, or scientific understanding |
Scientific validity | Uses accepted scientific methods likely to produce a valid answer to the study question |
Fair subject selection | Recruitment and eligibility of subjects are based on science and risk, not vulnerability or privilege |
Favorable risk/benefit ratio | Risks are minimized to the extent possible within the scientific objectives of the study; potential benefits are maximized Risks to subjects are justified by the sum of benefits to the subjects and to society |
Independent review | Study design and procedures must be reviewed and evaluated by individuals who are unaffiliated with the research |
Informed consent | Information must be provided to subjects about the nature of the research, benefits, risks, and alternatives; subjects must be able to make voluntary and reasoned decisions about enrolling In pediatric research, principles of surrogate permission and assent are applicable |
Respect for subjects | Investigators must: Protect subjects’ welfare Safeguard subjects’ privacy Allow withdrawal from the research Inform subjects of newly acquired information about risks, benefits, or alternatives Notify subjects of study results |
Special Requirements for Research in Children
In 1977, the Commission addressed the ethical issues surrounding the use of children as research subjects (National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research 1977). It recognized children as a particularly vulnerable population whose interests warrant additional safeguards yet acknowledged the tension between protecting children and excluding them from research altogether, precluding them from reaping the benefits of research. Shortly thereafter, the Code of Federal Regulations added Subpart D, outlining categories of research involving children that are permissible (Office for Protection from Research Risks 1983).
For nearly all research, parental permission is required, and the process “should be identical to that of informed consent” (Diekema 2006). In addition, the IRB determines when assent is required based on child participants’ capacity to provide it, using factors such as age, maturity, and psychological state. There is consensus that a capable child’s dissent ought to be respected, with one exception: when the research offers the prospect of direct benefit and is unavailable outside of the research context (Office for Protection from Research Risks 1983).
Prominent Ethical Dilemmas in Research
Phase I Trials
Case Vignette
A 7-year-old boy has recurrent and refractory metastatic rhabdomyosarcoma. Despite radiation and numerous chemotherapy regimens, his disease is progressing. You meet with the patient and his parents to discuss involvement of pediatric hospice services to optimize palliative care. During the meeting, the patient’s parents ask whether their son might be eligible for a new phase I trial opening in another state.
The goal of a typical phase I trial is to document the safety of a new intervention, most commonly by determining the dose-limiting toxicity (DLT) and maximum tolerated dose (MTD) of a novel drug. Critics contend that a phase I trial’s objective is to learn about safety, without therapeutic intent, raising the important question – can phase I trials offer the prospect of benefit to a child with cancer? (Miller and Joffe 2008; Agrawal and Emanuel 2003). By virtue of the animal data required to justify a phase I trial, the drug must show promise for patient subjects. And indeed, some studies suggest approximately 10 % of subjects have a partial or complete response (Lee et al. 2005). Moreover, many investigators insist that participants receive psychological benefits by contributing to future knowledge and by maintaining hope (Kodish et al. 1992). Once we accept that a trial offers the prospect of direct benefit, it must satisfy other regulatory criteria: (1) the risks of exposing a child to a novel intervention is justified by the anticipated benefit, and (2) the relation of anticipated benefit to risk is at least as favorable as that available with alternative approaches (Office for Protection from Research Risks 1983).
The final ethical concern we will address is that of informed consent. Critics contend that parents’ desperation for a cure makes them vulnerable. Available data demonstrates that participants enroll in phase I trials largely for personal benefit (Daugherty et al. 2000), that they overestimate the likelihood of benefit (Miller 2000), and that they have a poor understanding of the primary dose-finding objective of these studies (Cousino et al. 2012). While there is a dearth of quality data to attribute this to inadequate disclosure or instead to optimism, it is clear that a requisite component of any phase I trial is a transparent and forthright consent process.
Randomized Controlled Trials
Case Vignette
A 17-year-old young man recently diagnosed with a testicular tumor, and his family is considering a clinical trial in which subjects are randomly assigned to receive either standard chemotherapy or an alternate chemotherapy regimen that may have less toxicity. The patient is frustrated because he prefers the experimental regimen, but nobody can ensure this will be his regimen. He asks “why should I let a computer make decisions for me?”
Randomized controlled trials (RCTs) are the gold standard of biomedical research, allowing investigators to directly compare two interventions while controlling for factors that confound data generated by other study designs. In a typical oncology RCT, each subject is assigned, by chance, to one of two study groups. Subjects in the control group receive standard therapy, while those in the intervention group receive the treatment of interest. The classic ethical justification for such studies revolves around the concept of equipoise. Equipoise refers to “a state of professional uncertainty about [the] relative therapeutic merits” of the two interventions being studied (Miller and Joffe 2011). Put simply, if one of the two options is known to be better, we cannot justify randomly condemning half of the study population to receive the lesser therapy. Moreover, there is no need to conduct the study if we are certain which treatment is better. Conversely, most investigational therapies do not make it to a RCT unless there is evidence to suggest efficacy, so for a disease with poor therapeutic options, the presence of promising preliminary data might move expert opinion in favor of the novel drug, despite the lack of rigorous supporting data.
There are a host of criticisms of the concept of equipoise, and its use as the rationale for the appropriateness of RCTs (Miller and Brody 2003). Firstly, most patients expect their physician to weigh evidence, patient characteristics, and physician experience to determine the best therapeutic option. Equipoise assumes that patients will accept having that decision randomly assigned instead. Additionally, patients themselves may have a preference between treatments, even if the medical community does not. Thirdly, once a study begins, there may reach a point at which preliminary data favors one treatment over another. At this point equipoise, by definition, would be disturbed, and continuing the trial would be considered unethical. Data-monitoring committees are a part of most RCTs, and based on interim, data must either allow a study to continue or halt the study based on predefined stopping rules.
Therapeutic Misconception
Case Vignette
The parents of a 12-year-old female with newly diagnosed leukemia are considering whether to enroll their daughter in a clinical trial. As you meet with them, the patient’s mother says “I think we should do the trial. The doctors would not have offered it if they did not think it was the best thing for our daughter.”
The National Bioethics Advisory Commission defines the therapeutic misconception as “the belief that the purpose of a clinical trial is to benefit the individual patient rather than to gather data for the purpose of contributing to scientific knowledge” (National Bioethics Advisory Commission 2001). This concept is similar to the discussion of informed consent in phase I trials; however, it is also pertinent to RCTs such as the one described above. Despite clinicians’ best intentions, a significant percent of parents choosing the RCT will mistakenly believe that the clinician will select the treatment based on her judgment of which option is the best fit for the child (Kodish et al. 2004). There is increasing evidence that this type of misunderstanding is widespread, leading to calls for new approaches to trial enrollment, such as preventing clinicians from being the ones to present study details and offer enrollment (Eder et al. 2007; Appelbaum et al. 2012; Flory and Emanuel 2004).
Ethical Issues at the End of Life
Decision-Making about Life-Sustaining Treatments
When approaching decisions about life-sustaining therapies, parents of dying children often prioritize quality of life, likelihood of improvement, and perceptions of their child’s pain (Meyer et al. 2002). Similarly, physicians’ recommendations are guided by patient preferences about life support and prognostic factors, such as the likelihood of survival and the likely functional outcome (Cook et al. 2003). To support health-care providers who counsel families of patients who are dying, recommendations regarding limitation of life-sustaining treatments were advanced as early as 1983 by the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research 1983). Published guidelines of this kind also reflect lessons learned through paradigm cases (Table 17.3).
Table 17.3
Paradigm cases involving life-sustaining treatment decisions
Case | Facts of the case | Legal outcome | Relevance for ethical decision-making |
|---|---|---|---|
Baby Doe | Baby Doe was an infant with Down syndrome and tracheoesophageal fistula Parents declined corrective surgery stating that he would never achieve a “minimally acceptable quality of life” New federal regulations were proposed to protect infants with disabilities from having care withheld | Baby Doe law passed requiring all states to create a regulatory system to investigate cases where medically indicated treatment is withheld from handicapped infants | Relevance limited by the use of ambiguous terms not easily applied to actual cases Directed at state agencies, not individual health-care providers Not useful to clinicians making decisions regarding infants at the end of life |
Baby K | Baby K was an infant with anencephaly Mother demanded mechanical ventilation during multiple hospitalizations Clinicians refused, stating that mechanical ventilation could not reverse her malformation and therefore was not indicated | A federal appeals court ruled in favor of Baby K’s mother, arguing that the hospital was required to provide any emergent care, according to the Emergency Medical Treatment and Active Labor Act (EMTALA) | The court did not opine as to how futility enters into medical decisions Decision based on EMTALA rather than on the merits of the futility argument Case does not answer the key question: Can patients and families demand clinical care of providers who believe it will not be beneficial? |
Karen Ann Quinlan 1975, New Jersey (McFadden 1985) | Karen Ann Quinlan fell into a coma due to a combination of alcohol and sedative medications Parents requested the ventilator keeping her alive be discontinued Physicians refused the family’s request to let her die, a decision supported in the Superior Court of Morristown, N.J. Did not die for 9 more years when the ventilator was ultimately stopped | After a long legal battle, the Supreme Court decided 7–0 to support the parents in March 1976 Court justified their decision based on the patients right to privacy Stated that the patient’s father, not physicians, should make decisions of this kind | Provides an ethical argument for unwanted life-sustaining technologies to be considered unethical if requests to discontinue are not honored The clear medical assessment that recovery for the patient was not possible was essential to the case Decision-making absent clear prognostic data is even more challenging |
Nancy Cruzan | Cruzan was an adult patient involved in an automobile accident which left her in a “persistent vegetative state” She was sustained for 7 years with gastrostomy tube feedings | In a 5–4 decision, the Supreme Court ruled there was not enough evidence that Cruzan would have wanted the feeding tube removed | Parents and guardians must act in the best interests of children and incompetent adults Whether a particular intervention indeed reflects best interests can be controversial The US Supreme Court stated that there is no legal difference between withholding and withdrawing life-sustaining therapies |
Sedatives and Analgesics in the Care of the Dying
Health-care providers have an undeniable moral duty to treat the pain and suffering of terminally ill patients. Yet this imperative may also raise ethical questions if treating the pain and suffering could possibly hasten a patient’s death. Although empirical evidence suggests that the use of opioids and sedatives to relieve symptoms at the end of life in no way shortens life, theoretical concern remains (Macauley 2012; López-Saca et al. 2013). The ethical principle relevant to this question is the Doctrine of Double Effect (May 1978). The Doctrine states that when an action has two effects, one of which is inherently good and the other inherently bad, it can be justified if certain conditions are met:
1.
The action in itself must be good or at least morally indifferent.
2.
The clinician must intend only the good effect and not the bad effect. For example, the clinician must intend only the relief of the patient’s suffering by administering an opioid medication. The hastening of death due to respiratory depression may be foreseeable, but is not the clinician’s objective.
3.
The bad effect cannot be a means to the good effect. For example, the clinician may not administer a poison instead of morphine because doing so allows the bad effect (death) to be the means to the good effect (relief from suffering).
4.
The good intended must outweigh the potential bad outcome.
The moral reasoning outlined above remains controversial for a few reasons. First, according to the Doctrine, the moral justification for a clinician’s practice hinges on his intention, something that is impossible to know with certainty. In addition, critics believe that the only morally relevant consideration is the informed consent of the patient (or appropriate proxy), which is not explicitly considered in the Doctrine (Quill et al. 1997; Truog et al. 2012).
Medical Futility
Questions of futility arise in cancer medicine when patients or family members request care, such as chemotherapy, that is unlikely to be beneficial. Such requests may be motivated by unrealistic expectations of benefit if, for example, the likelihood that a treatment will improve symptoms or slow the pace of the disease is overestimated. In addition, requests for futile treatments may occur when an individual does not want to be viewed as “giving up” or “losing hope” (Khatcheressian et al. 2008). Disagreements between clinicians, patients, and family members about whether a particular treatment should be offered or continued may result in conflict. Resolving these conflicts hinges on high-quality communication, careful and honest counseling, and compassion on the part of all involved.
The concept and definition of futility has been debated for decades, most publicly in the Baby K case (Table 17.3). In general, proposed definitions lack an empirical basis, expert consensus, or societal endorsement (Burns and Truog 2007). Seeking a definition implies futility in an objective concept, while in reality, controversy around futility often centers on subjective preferences and values. Whether a treatment is valuable, what constitutes benefit, and whether preserving life is “better” than allowing death will remain the subject of ongoing debate.
Guidelines for Decision-Making
How should clinicians approach decisions for and with patients approaching the end of life (Fig. 17.1) In 1983 the President’s Commission proposed a useful and frequently cited construct for defining the best interests of a child approaching the end of life (President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research 1983). With these five considerations in mind, the clinician places a proposed treatment or intervention into one of three categories to define the potential benefit for the dying patient. The Commission advises on the best course of action for the patient, considering both the clinician assessment above and the preferences of the patients and parents. In general, when parents prefer to forego life-sustaining therapies that the clinician considers to have ambiguous/uncertain benefit or to be clearly not beneficial, then the clinician should withdraw life-sustaining therapy (Fig. 17.1).
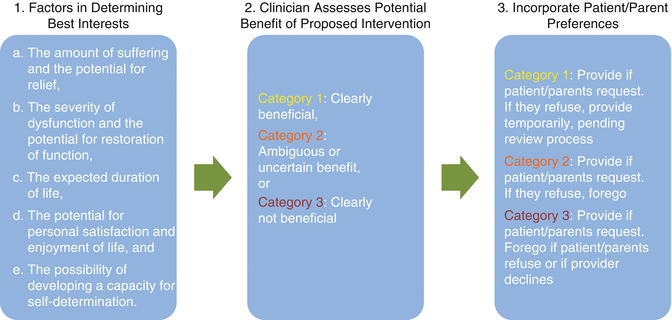
Fig. 17.1
Approach to decision-making at the end of life
Prominent Ethical Dilemmas at the End of Life
Palliative Sedation
Case Vignette
A 10-year-old with a relapsed and refractory tumor of the brain experiences intractable seizures. The patient and family are intensely disturbed by the seizures and express their primary goal of halting the seizure activity. Doing so requires large doses of sedating medications such that the patient is no longer conscious. The medical team plans to decrease the medications in 48 h to “see how the patient will do,” but the family requests the medications continue, stating, “this is the only way our son can have peace.”
A rarely used approach to sedation and analgesia at the end of life is palliative sedation, wherein a child must be sedated to the point of unconsciousness in order to be comfortable (Truog et al. 1992; Cowan and Walsh 2001; Postovsky et al. 2007). This practice should be considered, with consent, when all other methods of controlling the patient’s suffering have failed (National Ethics Committee 2006). After the patient loses consciousness, hydration and nutrition stop and no attempt is typically made to restore the patient to consciousness. Patients typically die after several days from dehydration or a complication of the treatment.
The US Supreme Court implicitly endorsed palliative sedation, citing the technique as an alternative to physician-assisted suicide that could, in theory, assure that no patient should die with “untreatable” pain (McStay 2003). At least in part because of this legal endorsement, palliative sedation has become more widely practiced, although its ethical justification remains controversial (Schüklenk et al. 2011; Kiman et al. 2011; Goldstein et al. 2012).
Withholding or Withdrawal of Medical Nutrition and Hydration
Case Vignette
A 14-year-old female has a large intra-abdominal tumor compressing her bowel. Despite numerous chemotherapy regimens, the tumor has progressed. The patient is no longer receiving cancer-directed therapy and both she and her family realize that she will likely die from this cancer. The patient is admitted to the hospital with vomiting and inability to tolerate her nasogastric feeds. The tumor has caused an obstruction in her bowel. The team recommends surgery to alleviate the obstruction, but the patient requests that nasogastric feeds simply be discontinued.
The decision to withhold or withdraw medical nutrition and hydration should be made after weighing the burdens and benefits of therapy for that patient. Such decisions are morally equivalent to those pertaining to mechanical ventilation and other life-sustaining interventions (Levi 2003; Diekema and Botkin 2009; Nelson 1987); however, they can be controversial since many consider nutrition and hydration to be basic supports for ongoing life, without which human dignity is compromised. Some argue that decisions to forego medical nutrition and hydration can constitute patient abandonment. Despite these concerns, courts have ruled that they are medical interventions and may be discontinued on the same grounds as any other medical treatment (Table 17.3).
Ethical Issues in Genetic Testing and Diagnosis
Rapid advances in biomedical technology have increased the accessibility of genetic testing. These evaluations are now an intrinsic component of clinical practice, and in the research context, genetic testing is even more prevalent. Although the intent of such testing is to elucidate genetic mutations underlying a particular tumor, incidental findings may have wider implications for the patient and family, raising new ethical questions (Presidential Commission for the Study of Bioethical Issues 2012).
Information gleaned from genetic testing may be of pivotal importance to the care of a child with cancer. Genetic tests performed on the tumor can reveal details that may influence treatment decisions. In many cases, these tests may be considered standard of care and new research opportunities related to this testing are increasingly common. Other genetic testing performed on the patient may reveal whether a cancer is likely a random event or possibly part of an inherited predisposition to cancer. In this way, results of testing performed for a single patient may be of consequence for that patient’s family members, many of whom are not included in decision-making about the tests. Another complexity is that the data obtained from any of these genetic tests may be difficult to interpret. Results are often of unclear significance, stated in terms of risk or probability of affecting the patient in a meaningful way. Because both risk and probability can be challenging for patients and clinicians to understand clearly, these results may generate confusion and distress, an additional psychological risk of genetic testing.
Prominent Ethical Dilemmas in Genetics and Genomics
Cancer Predisposition Testing



