25 Endocrine and Metabolic Disorders
 Anatomy and Physiology
Anatomy and Physiology Pathophysiology
Pathophysiology
Many metabolic diseases are caused by inborn errors of metabolism. An alteration in genetic constitution results in disrupted biochemical functioning. For example, in children with phenylketonuria (PKU, a deficiency of the enzyme phenylalanine hydroxylase), the essential amino acid phenylalanine accumulates, resulting in mental retardation if not treated within the first weeks of life. Type 1 diabetes mellitus is an example of an acquired immune-mediated metabolic disease. In diabetes, a reduction in insulin production or deficiency of its action results in abnormal metabolism of carbohydrate, protein, and fat.
 Assessment
Assessment
History
• What is the child’s growth pattern since birth?
• Has there been a recent alteration in growth pattern?
• Is the child taking any medications that could affect endocrine or metabolic function?
• Have there been signs or symptoms of endocrine or metabolic dysfunction?
• Was there maternal exposure to radioiodine, goitrogens, or iodine medication during pregnancy?
• When did the child first show signs of sexual development?
• What is the child’s diet and exercise history?
• Is there a family history of endocrine, autoimmune, or metabolic disorders?
• Does the child have unusual odors, recurrent vomiting, or unexplained lethargy?
Physical Examination
A detailed examination should include the following:
• Measure stature. Supine length is preferred for children younger than 2 years old. Use a stadiometer for children older than 2 or 3 years. Plot height, weight, and head circumference on a standardized growth chart appropriate to the child’s age and gender. In addition, growth charts are available for children with certain genetic conditions, such as Down and Turner syndromes, and should be used to assess growth patterns of children with these conditions (see Appendix B). Serial measurements are critical to assess growth patterns over time.
• Check for proportionate appearance. Measure sitting and standing heights for upper to lower segment ratio (see Chapter 32).
• Assess height age (the age corresponding to the child’s height when plotted at the 50th percentile on a growth chart) and growth velocity (linear growth in centimeters or inches over the past year).
• Inspect the child’s genitalia for signs of either normal or ambiguous genitalia.
• Identify the stage of sexual development using Tanner staging (see Chapter 8).
• Note facial, axillary, and pubic hair for presence, distribution, and texture.
• Examine the skin for presence of striae and acanthosis nigricans (see Color Plate) of the neck, axilla, breast, knuckles, and skinfolds.
• Palpate the neck for thyroid gland symmetry and size, noting enlargement or presence of nodules.
• Examine for presence of dysmorphic features.
• Examine the abdomen noting any organomegaly.
 Management Strategies
Management Strategies
General Measures
• Family, school, peer, and emotional adjustment
• Body image, self-esteem, and social competence
• Disease understanding, acceptance, and self-care
Genetic Counseling
Genetic counseling is often necessary. Implications are significant for the family of a child with endocrine or metabolic disorders that are genetically linked (see Chapter 40).
Medications
Pharmacologic therapy, including hormone replacement, whether temporary or lifelong is often essential for management of these disorders. Medications may be administered via injection, creating distress in both the child and caregiver. Short, clear instructions about medications are important; how much to give, when and how to administer, possible side effects, and when to make adjustments in medication are key messages to convey.
 Growth Disorders
Growth Disorders

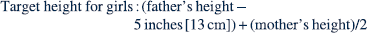
Growth disorders may be classified as primary or secondary. Primary growth disorders include skeletal dysplasias, chromosomal abnormalities (e.g., Turner syndrome), and genetic short stature. Secondary growth disorders may result from undernutrition, chronic disease, endocrine disorder, and idiopathic (constitutional) growth delay [CGD]) (Box 25-1). The following discussion focuses on growth hormone deficiency (GHD) and CGD (Table 25-1).
BOX 25-1 Classification of Growth Retardation





IGF-1, Insulin-like growth factor 1; SHOX, short stature homeobox.
TABLE 25-1 Short Stature: Characteristics of Growth Hormone Deficiency and Constitutional Growth Delay in Children
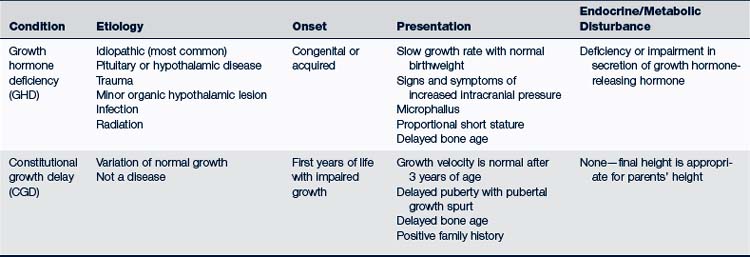
Growth Hormone Deficiency
Epidemiology
Estimates of the incidence of idiopathic GHD vary; an epidemiologic study conducted in Denmark reported an incidence of child-onset GHD of 2.58/100,000 for boys and 1.7/100,000 for girls (Stochholm et al, 2006).
Clinical Findings
History
A history obtained to evaluate the short or slowly growing child should include:





Physical Examination
Physical examination of the short or slow-growing child should include:
• Identification of clinical clues to chronic illness or dysmorphic syndrome (e.g., childlike facies with large, prominent forehead)
• Evaluation of the fundi for signs of increased intracranial pressure
• Palpation of the thyroid gland for the presence of a goiter
• Evaluation of the stage of puberty
• Measurement of body proportions including arm span, height and upper-to-lower (U/L) body segment ratio to exclude a skeletal dysplasia (dwarfing condition). Interpretation of the U/L body segment ratio is dependent on the age of the child. At birth the U/L ratio is approximately 1.7:1; at 3 years of age it is approximately 1.3:1; and at 7 years and older approximately 1:1 (Keane, 2007).
Diagnostic Tests
• Complete blood count (CBC) and erythrocyte sedimentation rate (ESR)
• Screening for gastrointestinal (GI) illness when appropriate (e.g., celiac disease screening [serum immunoglobulin A (IgA) and transglutaminase], irritable bowel disease, stool for ova and parasites)
• Growth factors (IGF-1 and IGFBP-3)
• Thyroid function tests: Free T4 and TSH should be obtained to exclude both pituitary TSH deficiency and primary hypothyroidism
• Bone age x-ray of left wrist and hand
• Karyotype to rule out Turner syndrome in girls. Turner syndrome occurs in approximately 1:1500 to 1:2500 girls. Girls with Turner mosaicism may not manifest the typical clinical findings of Turner syndrome (e.g., cubitus valgus, webbing of the neck) thus highlighting the importance of karyotyping all females presenting with short stature (Loscalzo, 2008) (see Chapter 40).
• Measurement of GH production may be necessary. Because secretion of GH is pulsatile, random serum measurement of the hormone is inadequate; stimulation testing using agents such as arginine, levodopa (L-dopa), clonidine, and/or glucagon is needed to accurately assess GH production.
Differential Diagnosis
Individual children with short stature may not fit nicely into a single category, but may have multiple factors contributing to their stature. Many chronic illnesses can slow linear growth, likely through a variety of mechanisms including malnutrition, acidosis, anorexia, and deficiencies of minerals (e.g., zinc and iron) and vitamins necessary for growth (Box 25-2). Typically children with poor growth as a result of chronic illness are underweight for height; their weight gain slows prior to growth deceleration. Thyroid hormones are essential to growth during childhood; sex steroids are important for normal growth during the pubertal growth spurt. Deficiency of these hormones is characterized by subnormal growth velocity, normal to increased weight for height, and delay in bone age.








Management
Children should be referred to a pediatric endocrinologist if hypothyroidism, low IGF-1 and IGFBP-3 or other hormone deficiency is confirmed, or for unexplained persistent slow growth without evidence of chronic illness. The U.S. Food and Drug Administration (FDA) has approved a number of indications for GH therapy (Box 25-3) (Schwenk, 2006). GH dosing is based on a child’s body weight with doses ranging from 0.15 to 0.30 mg/kg/wk. Response to GH therapy is greater for children receiving daily injections compared with those receiving three injections per week. During the first year of therapy, growth velocity may exceed normal growth rates as much as fourfold. Reported side effects of GH include glucose intolerance, pseudotumor cerebri, edema, growth of nevi, slipped capital femoral epiphyses, and scoliosis (Schwenk, 2006). The cost of GH therapy may present an economic burden to the family and referral to a social worker to assist in finding financial support may be appropriate.


Constitutional Growth Delay
Clinical Findings
History
The history may include the following:
• Normal length and weight at birth
• Slowed linear growth between 1 to 3 years of age and then normal growth velocity; normal height velocity is the most critical factor in diagnosing CGD
• Height at or slightly below the third percentile on standardized growth charts
• Delayed pubertal development
• History of similar growth patterns in other family members
Growth Excess
• Primary skeletal abnormalities such as Marfan syndrome, Klinefelter syndrome, and other overgrowth syndromes
• Overnutrition that advances the bone age and the timing of puberty. In these children, weight gain occurs first, and weight percentile is farther above the growth curve than height percentile.
• Excess adrenal androgens or gonadal steroids. These children have physical examination findings of early puberty.
 Pubertal Disorders
Pubertal Disorders
The physical changes of puberty occur in response to production of sex steroids by the ovaries or testes (see Chapter 8). Hypothalamic gonadotropin-releasing hormone (GnRH) regulates the release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the pituitary gland, which in turn stimulates gonadal hormone secretion.
Precocious Puberty
Description
True precocious puberty refers to the onset of multiple features of puberty earlier than the normal range. Features may include accelerated linear growth, breast development or penile enlargement, and pubic hair development. Depending on the duration of symptoms, the bone age may be advanced. Precocious puberty can be divided into two broad categories: central, gonadotropin dependent; or peripheral, gonadotropin independent (Box 25-4). Prolonged exposure to exogenous sex hormones (mother’s birth control pills or father’s topical testosterone) (Aksglaede et al, 2006) and exposure to chemicals that disrupt endocrine function (see Chapter 41) can cause precocious puberty (Cesario and Hughes, 2007).





Clinical Findings
Children who present with features of puberty at a younger age than normal should have an evaluation as to the etiology. Children who start to develop signs of puberty at the early end of the normal range should be evaluated if they have rapid progression of pubertal signs resulting in a bone age more than 2 years ahead of chronologic age, or new CNS-related findings (e.g., headaches, seizures, focal neurologic defects) (Kaplowitz, 2009).
Physical Examination
Physical examination should include:
• Assessment of stature and growth velocity
• Description of the child’s Tanner stage
• Breast development should be evaluated by palpation rather than inspection to differentiate between the presence of true breast tissue versus fat deposition (Rosenfield et al, 2009)
• Pubic and axillary hair (girls)
• Penile length, testicular volume, and pubic and axillary hair (boys) (see Chapter 8)
Diagnostic Tests
• Premature thelarche: no laboratory studies are necessary in the infant or toddler girl unless she has other features of true puberty or continued increase in breast size
• Premature adrenarche: serum 17-hydroxyprogesterone (17-OHP) to exclude CAH and a 24-hour urine collection for 17-ketosteroids or imaging of the adrenal glands to exclude an adrenal tumor
• Isolated menarche: thyroid function tests to exclude primary hypothyroidism, and pelvic ultrasound to rule out the presence of an ovarian cyst or pelvic tumor
Diagnostic tests for children with true precocious puberty include:
• LH, FSH, and estradiol or testosterone (use a laboratory with a sensitive assay that will detect early pubertal values at the lower end of the range)
• If LH and FSH are high (in pubertal range: indication of central etiology), an MRI is indicated to exclude CNS tumor.
• If LH and FSH are low (in prepubertal range: indication of peripheral puberty), complete a GnRH stimulation test to distinguish central from peripheral puberty.



Delayed Puberty
Epidemiology
Any chronic condition that delays the bone age may cause delayed puberty because the timing of puberty correlates better with bone age than chronologic age (Box 25-5). In addition, failure of any part of the hypothalamic-pituitary-gonadal axis may also delay puberty (Box 25-6). The most common cause of delayed puberty is CGD (Louis et al, 2008).

Clinical Findings
History and Physical Examination
History and physical examination should focus on clinical clues indicating a chronic illness, symptoms or signs of hypothyroidism, prior history of CNS insult, or new CNS symptoms suggesting hypopituitarism. Review of systems should include questions about pattern of growth, especially growth velocity, sense of smell, and galactorrhea.
Diagnostic Tests
Laboratory investigation should include:
 Adrenal Disorders
Adrenal Disorders
Anatomy and Physiology
Adrenal gland steroid production is under the control of the hypothalamic-pituitary axis. The hypothalamus secretes corticotropin-releasing hormone (CRH) in a pulsatile fashion, which stimulates production and secretion of adrenocorticotropic hormone (ACTH) by the pituitary gland. ACTH regulates adrenal glucocorticoid (cortisol) and androgen production. Cortisol is produced in a series of enzymatic steps (Fig. 25-1) and is highest in the morning, low in the afternoon and evening, and lowest at midnight. Secreted in response to hypoglycemia, hypotension, pain, or other stressful events, cortisol has negative feedback on the synthesis and secretion of CRH, vasopressin, and ACTH.
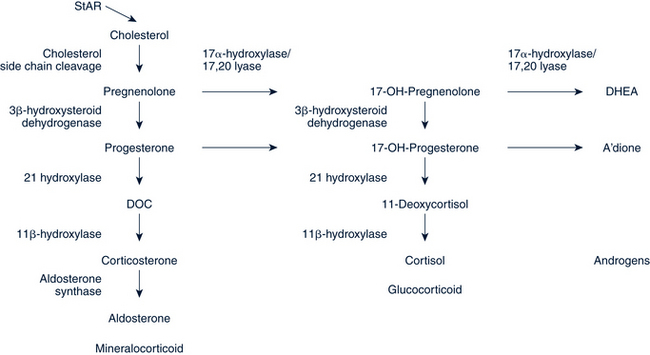
Adrenal Insufficiency
Epidemiology
Primary adrenal insufficiency may be due to an inability to produce cortisol secondary to an enzyme defect in the adrenal steroid pathway (CAH), hypoplasia of the adrenal gland, or an acquired defect (Box 25-7). Lesions of the hypothalamus or pituitary lead to secondary adrenal insufficiency. Suppression of the hypothalamic-pituitary-adrenal axis secondary to steroid use can also lead to adrenal insufficiency. Infants born extremely prematurely (24 to 28 weeks’ gestation) sometimes demonstrate symptoms of adrenal insufficiency because of immaturity of the hypothalamic-pituitary-adrenal axis.









Clinical Findings
History
Symptoms of cortisol deficiency include a history of:
Symptoms of aldosterone deficiency include:
Diagnostic Tests
• Serum glucose (hypoglycemia)
• Blood gases and bicarbonate (for metabolic acidosis)
• Electrolytes (low sodium, elevated potassium with aldosterone deficiency)
• Serum cortisol (a cortisol value greater than 20 mcg/dL indicates adrenal sufficiency. A value lower than that must be interpreted in the clinical context in which the sample was drawn. Often an ACTH stimulation test, performed in collaboration with a pediatric endocrinologist, is needed to conclusively diagnose both primary and secondary adrenal insufficiency).
• Serum ACTH (elevated in primary adrenal insufficiency)
• Serum 17-OHP (diagnostic in children with suspected CAH caused by 21-OH deficiency)
• Serum renin level (elevated with aldosterone deficiency)
Management
Long-term therapy of CAH includes oral hydrocortisone in replacement doses of 8 to 10 mg/m2 (8 to 10 mg per square meter of body surface) in children with ACTH deficiency or primary adrenal insufficiency. Children with CAH tend to have higher hydrocortisone needs. If present, aldosterone deficiency must be treated with fludrocortisone acetate. Treatment of CAH requires a fine balancing act to replace steroids, thereby preventing androgen overproduction. Excess steroid intake can lead to delayed growth; not enough steroids contribute to rapid bone age growth and ultimate short stature. Individual treatment plans are essential to meet the specific needs of individual children. The primary care provider should be familiar with the medical endocrinology treatment plan and reinforce it at routine well- and sick-child visits.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


