Chapter 52 Electrolyte and Acid-Base Disorders
52.1 Composition of Body Fluids
Total Body Water
Water is the most plentiful constituent of the human body. Total body water (TBW) as a percentage of body weight varies with age (Web  Fig. 52-1). The fetus has very high TBW, which gradually decreases to approximately 75% of birthweight for a term infant. Premature infants have higher TBW than term infants. During the 1st yr of life, TBW decreases to approximately 60% of body weight and basically remains at this level until puberty. At puberty, the fat content of females increases more than that of males, who acquire more muscle mass than females. Because fat has very low water content and muscle has high water content, by the end of puberty TBW in males remains at 60%, but TBW in females decreases to approximately 50% of body weight. The high fat content in overweight children causes a decrease in TBW as a percentage of body weight. During dehydration, TBW decreases and, thus, is a smaller percentage of body weight.
Fig. 52-1). The fetus has very high TBW, which gradually decreases to approximately 75% of birthweight for a term infant. Premature infants have higher TBW than term infants. During the 1st yr of life, TBW decreases to approximately 60% of body weight and basically remains at this level until puberty. At puberty, the fat content of females increases more than that of males, who acquire more muscle mass than females. Because fat has very low water content and muscle has high water content, by the end of puberty TBW in males remains at 60%, but TBW in females decreases to approximately 50% of body weight. The high fat content in overweight children causes a decrease in TBW as a percentage of body weight. During dehydration, TBW decreases and, thus, is a smaller percentage of body weight.
Fluid Compartments
TBW is divided between 2 main compartments: intracellular fluid (ICF) and extracellular fluid (ECF). In the fetus and newborn, the ECF volume is larger than the ICF volume (see Web Fig. 52-1). The normal postnatal diuresis causes an immediate decrease in the ECF volume. This is followed by continued expansion of the ICF volume, which results from cellular growth. By 1 yr of age, the ratio of the ICF volume to the ECF volume approaches adult levels. The ECF volume is approximately 20-25% of body weight, and the ICF volume is approximately 30-40% of body weight, close to twice the ECF volume (Web Fig. 52-2). With puberty, the increased muscle mass of males causes them to have a higher ICF volume than females. There is no significant difference in the ECF volume between postpubertal females and males.
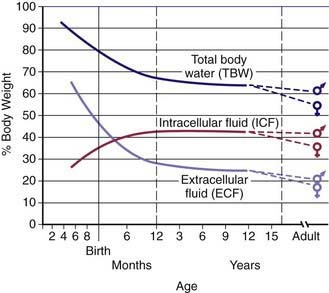
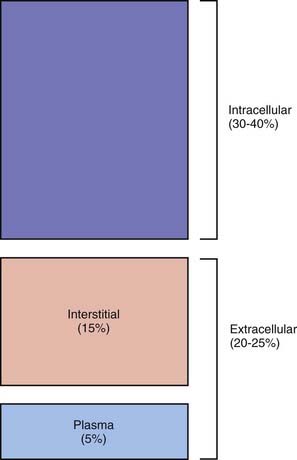
Web Figure 52-2 Compartments of total body water, expressed as percentages of body weight, in an older child or adult.
The ECF is further divided into the plasma water and the interstitial fluid (see Web Fig. 52-2). The plasma water is 5% of body weight. The blood volume, given a hematocrit of 40%, is usually 8% of body weight, although it is higher in newborns and young infants; in premature newborns, it is approximately 10% of body weight. The volume of plasma water can be altered by pathologic conditions, including dehydration, anemia, polycythemia, heart failure, abnormal plasma osmolality, and hypoalbuminemia. The interstitial fluid, normally 15% of body weight, can increase dramatically in diseases associated with edema, such as heart failure, protein-losing enteropathy, liver failure, nephrotic syndrome, and sepsis. An increase in interstitial fluid also occurs in patients with ascites or pleural effusions.
Electrolyte Composition
The composition of the solutes in the ICF and ECF are very different (Web Fig. 52-3). Sodium and chloride are the dominant cation and anion, respectively, in the ECF. The sodium and chloride concentrations in the ICF are much lower. Potassium is the most abundant cation in the ICF, and its concentration within the cells is approximately 30 times higher than in the ECF. Proteins, organic anions, and phosphate are the most plentiful anions in the ICF. The dissimilarity between the anions in the ICF and the ECF is largely determined by the presence of intracellular molecules that do not cross the cell membrane, the barrier separating the ECF and the ICF. In contrast, the difference in the distribution of cations—sodium and potassium—is due to the activity of the Na+,K+-ATPase pump, which uses cellular energy to actively extrude sodium from cells and move potassium into cells. The chemical gradient between the intracellular potassium concentration and the extracellular potassium concentration creates the electrical gradient across the cell membrane. The concentration-dependent movement of potassium out of the cell makes the intracellular space negative relative to the extracellular space.
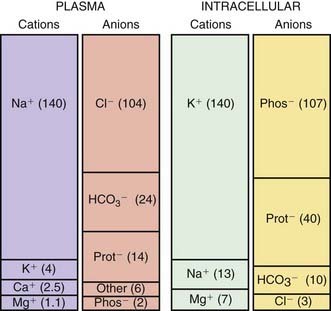
Web Figure 52-3 The concentrations of the major cations and anions in the intracellular space and the plasma, expressed in mEq/L.
Osmolality
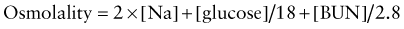
Glucose and blood urea nitrogen (BUN) are measured in mg/dL. Division of these values by 18 and 2.8, respectively, as shown, converts the units into mmol/L. Multiplication of the sodium value by 2 accounts for its accompanying anions, principally chloride and bicarbonate. The calculated osmolality is usually slightly lower than the measured osmolality.
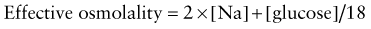
The effective osmolality (also called the tonicity) determines the osmotic force that is mediating the shift of water between the ECF and the ICF.
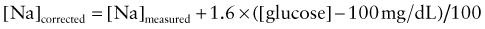
where [Na]measured = sodium concentration measured by the clinical laboratory and [Na]corrected = corrected sodium concentration (the sodium concentration if the glucose concentration were normal and its accompanying water moved back into the cells). The [Na]corrected is the more reliable indicator of the patient’s true ratio of total body sodium to TBW, the normal determinant of the sodium concentration.
Garibaldi BT, Cameron SJ, Choi M. Pseudohyponatremia in a patient with HIV and hepatitis C coinfection. J Gen Intern Med. 2008;23:202-205.
Nguyen MK, Ornekian V, Butch AW, et al. A new method for determining plasma water content: application in pseudohyponatremia. Am J Physiol Renal Physiol. 2007;292:F1652-F1656.
Pellegrino B, Parravani A, Cook L, et al. Ethylene glycol intoxication: disparate findings of immediate versus delayed presentation. W V Med J. 2006;102:32-34.
52.2 Regulation of Osmolality and Volume
Regulation of Osmolality
Osmoreceptors in the hypothalamus sense the plasma osmolality (Chapter 552). An elevated effective osmolality leads to secretion of antidiuretic hormone (ADH) by neurons in the supraoptic and paraventricular nuclei in the hypothalamus. The axons of these neurons terminate in the posterior pituitary. Circulating ADH binds to its V2 receptors in the collecting duct cells of the kidney, and, via the generation of cyclic adenosine monophosphate, causes insertion of water channels (aquaporin-2) into the renal collecting ducts. This produces increased permeability to water, permitting resorption of water into the hypertonic renal medulla. The end result is that the urine concentration increases and water excretion decreases. Urinary water losses cannot be completely eliminated because there is obligatory excretion of urinary solutes, such as urea and sodium. The regulation of ADH secretion is tightly linked to plasma osmolality, responses being detectable with a 1% change in the osmolality. ADH secretion virtually disappears when the plasma osmolality is low, allowing excretion of maximally dilute urine. The consequent loss of free water (water without sodium) corrects the plasma osmolality. ADH secretion is not an all-or-nothing response; there is a graded adjustment as the osmolality changes.
A number of conditions can limit the kidney’s ability to excrete adequate water to correct low plasma osmolality. In the syndrome of inappropriate antidiuretic hormone (SIADH), ADH continues to be produced despite a low plasma osmolality. In the presence of ADH, urinary dilution does not occur, and sufficient water is not excreted (Chapters 52.3 and 553).
The normal response to increased plasma osmolality is conservation of water by the kidney. In central diabetes insipidus, this does not occur because of an absence of ADH secretion (Chapter 552.1). Patients with nephrogenic diabetes insipidus have an inability to respond to ADH and produce dilute urine despite an increase in plasma osmolality (Chapters 52.3, 524, and 552).
Regulation of Volume
Sodium resorption occurs throughout the nephron (Chapter 522). Whereas the majority of filtered sodium is resorbed in the proximal tubule and the loop of Henle, the distal tubule and the collecting duct are the main sites for precise regulation of sodium balance. Approximately 65% of the filtered sodium is reclaimed in the proximal tubule, which is the major site for resorption of bicarbonate, glucose, phosphate, amino acids, and other substances that are filtered by the glomerulus. The transport of all these substances is linked to sodium resorption by cotransporters, or a sodium-hydrogen exchanger in the case of bicarbonate. This link is clinically important for bicarbonate and phosphate because their resorption parallels sodium resorption. In patients with metabolic alkalosis and volume depletion, correction of the metabolic alkalosis requires urinary loss of bicarbonate, but the volume depletion stimulates sodium and bicarbonate retention, preventing correction of the alkalosis. Volume expansion causes increased urinary losses of phosphate, even when there is phosphate depletion. Resorption of uric acid and urea occurs in the proximal tubule and increases when sodium retention increases. This arrangement accounts for the elevated uric acid and BUN measurements that often accompany dehydration, which is a stimulus for sodium retention in the proximal tubule. The cells of the proximal tubule are permeable to water; thus, water resorption in this segment parallels sodium resorption.
Volume depletion usually occurs when sodium losses exceed intake. The most common etiology in children is gastroenteritis. Excessive losses of sodium may also occur from the skin in children with burns, in sweat from patients with cystic fibrosis, or after vigorous exercise. Inadequate intake of sodium is uncommon except in neglect, in famine, or with an inappropriate choice of liquid diet in a child who cannot take solids. Urinary sodium wasting may occur in a range of renal diseases, from renal dysplasia to tubular disorders, such as Bartter syndrome. The neonate, especially if premature, has a mild impairment in the ability to conserve sodium. Iatrogenic renal sodium wasting takes place during diuretic therapy. Renal sodium loss occurs as a result of derangement in the normal regulatory systems. An absence of aldosterone, seen most commonly in children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency, causes sodium wasting (Chapter 570).
Ball SG. Vasopressin and disorders of water balance: the physiology and pathophysiology of vasopressin. Ann Clin Biochem. 2007;44:417-431.
Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19:1076-1078.
Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519-531.
Nielsen S, Kwon TH, Frokiaer J, et al. Regulation and dysregulation of aquaporins in water balance disorders. J Intern Med. 2007;261:53-64.
Schrier RW. Aquaporin-related disorders of water homeostasis. Drug News Perspect. 2007;20:447-453.
Schrier RW. Decreased effective blood volume in edematous disorders: what does this mean? J Am Soc Nephrol. 2007;18:2028-2031.
52.3 Sodium
Sodium Metabolism
Body Content and Physiologic Function
Sodium is the dominant cation of the ECF (see  Web Fig. 52-3), and it is the principal determinant of extracellular osmolality. Sodium is therefore necessary for the maintenance of intravascular volume. Less than 3% of sodium is intracellular. More than 40% of total body sodium is in bone; the remainder is in the interstitial and intravascular spaces. The low intracellular sodium concentration, approximately 10 mEq/L, is maintained by Na+,K+-ATPase, which exchanges intracellular sodium for extracellular potassium.
Web Fig. 52-3), and it is the principal determinant of extracellular osmolality. Sodium is therefore necessary for the maintenance of intravascular volume. Less than 3% of sodium is intracellular. More than 40% of total body sodium is in bone; the remainder is in the interstitial and intravascular spaces. The low intracellular sodium concentration, approximately 10 mEq/L, is maintained by Na+,K+-ATPase, which exchanges intracellular sodium for extracellular potassium.
Intake
Sodium is readily absorbed throughout the gastrointestinal tract. Mineralocorticoids increase sodium transport into the body, although this effect has limited clinical significance. The presence of glucose enhances sodium absorption owing to the presence of a cotransport system. This is the rationale for including sodium and glucose in oral rehydration solutions (Chapter 332).
Hypernatremia
Etiology and Pathophysiology
There are 3 basic mechanisms of hypernatremia (Table 52-1). Sodium intoxication is frequently iatrogenic in a hospital setting as a result of correction of metabolic acidosis with sodium bicarbonate. Baking soda, a putative home remedy for upset stomach, is another source of sodium bicarbonate; the hypernatremia is accompanied by a profound metabolic alkalosis. In hyperaldosteronism, there is renal retention of sodium and resultant hypertension; the hypernatremia is usually mild.
Table 52-1 CAUSES OF HYPERNATREMIA
EXCESSIVE SODIUM
WATER DEFICIT
WATER AND SODIUM DEFICITS
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
The classic causes of hypernatremia from a water deficit are nephrogenic and central diabetes insipidus (Chapters 524 and 552). Hypernatremia develops in diabetes insipidus only if the patient does not have access to water or cannot drink adequately because of immaturity, neurologic impairment, emesis, or anorexia. Infants are at high risk because of their inability to control their own water intake. Central diabetes insipidus and the genetic forms of nephrogenic diabetes insipidus typically cause massive urinary water losses and very dilute urine. The water losses are less dramatic, and the urine often has the same osmolality as plasma when nephrogenic diabetes insipidus is secondary to disease (obstructive uropathy, renal dysplasia, sickle cell disease).
When hypernatremia occurs in conditions with deficits of sodium and water, the water deficit exceeds the sodium deficit. This occurs only if the patient is unable to ingest adequate water. Diarrhea results in depletion of both sodium and water. Because diarrhea is hypotonic—typical sodium concentration of 35-65 mEq/L—water losses exceed sodium losses, potentially leading to hypernatremia. Most children with gastroenteritis do not have hypernatremia because they drink enough hypotonic fluid to compensate for stool water losses (Chapter 332). Fluids such as water, juice, and formula are more hypotonic than the stool losses, allowing correction of the water deficit and potentially even causing hyponatremia. Hypernatremia is most likely to occur in the child with diarrhea who has inadequate intake because of emesis, lack of access to water, or anorexia.
Clinical Manifestations
Most children with hypernatremia are dehydrated and show the typical clinical signs and symptoms (Chapter 54). Children with hypernatremic dehydration tend to have better preservation of intravascular volume because of the shift of water from the intracellular space to the extracellular space. This shift maintains blood pressure and urine output and allows hypernatremic infants to be less symptomatic initially and potentially to become more dehydrated before medical attention is sought. Breast-fed infants with hypernatremia are often profoundly dehydrated, with failure to thrive. Probably because of intracellular water loss, the pinched abdominal skin of a dehydrated, hypernatremic infant has a “doughy” feel.
Diagnosis
When there is isolated water loss, the signs of volume depletion are usually less severe initially because much of the loss is from the intracellular space. When pure water loss causes signs of dehydration, the hypernatremia and water deficit are usually severe. In the child with renal water loss, either central or nephrogenic diabetes insipidus, the urine is inappropriately dilute and urine volume is not low. The urine is maximally concentrated and urine volume is low if the losses are extrarenal or due to inadequate intake. With extrarenal causes of loss of water, the urine osmolality should be >1,000 mOsm/kg. When diabetes insipidus is suspected, the evaluation may include measurement of ADH and a water deprivation test, including a trial of desmopressin acetate (synthetic ADH analog) to differentiate between nephrogenic diabetes insipidus and central diabetes insipidus (Chapter 552.1). A water deprivation test is unnecessary if the patient has simultaneous documentation of hypernatremia and poorly concentrated urine (osmolality lower than that of plasma). In children with central diabetes insipidus, administration of desmopressin acetate increases the urine osmolality above the plasma osmolality, although maximum osmolality does not occur immediately because of the decreased osmolality of the renal medulla due to the chronic lack of ADH. In children with nephrogenic diabetes insipidus, there is no response to desmopressin acetate.
Treatment
As hypernatremia develops, the brain generates idiogenic osmoles to increase the intracellular osmolality and prevent the loss of brain water. This mechanism is not instantaneous and is most prominent when hypernatremia has developed gradually. If the serum sodium concentration is lowered rapidly, there is movement of water from the serum into the brain cells to equalize the osmolality in the 2 compartments (Fig. 52-1). The resultant brain swelling manifests as seizures or coma.
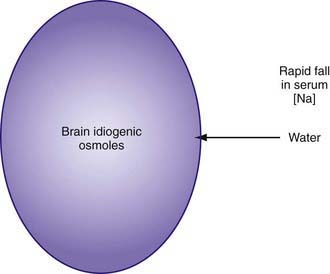
In the child with hypernatremic dehydration, as in any child with dehydration, the first priority is restoration of intravascular volume with isotonic fluid (Chapter 54). Normal saline is preferable to lactated Ringer solution because the lower sodium concentration of the latter can cause the serum sodium to decrease too rapidly, especially if multiple fluid boluses are given. Repeated boluses of normal saline (10-20 mL/kg) may be required to treat hypotension, tachycardia, and signs of poor perfusion (peripheral pulses, capillary refill time) (Chapters 54 and 64).
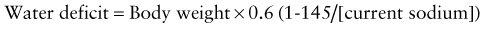
This calculation is equivalent to 3-4 mL of water per kg for each 1 mEq that the current sodium level exceeds 145 mEq. The utility of such formulas has never been proven in clinical practice. Most patients with hypernatremic dehydration do well with a fluid sodium concentration of approximately half-normal saline, but with a fluid rate that is only 20-30% greater than maintenance fluid. Use of this concentration prevents excessive delivery of free water and too rapid a decrease in the serum sodium level. Patients with pure water loss may require a more hypotonic fluid (0.2 normal saline). Excessive water and sodium losses may also need to be replaced. If signs or symptoms of volume depletion develop, the patient receives additional boluses of isotonic saline. Monitoring of the rate of decrease of the serum sodium concentration permits adjustment in the rate and sodium concentration of the fluid that the patient is receiving, avoiding overly rapid correction of the hypernatremia. Many patients with mild to moderate hypernatremic dehydration due to gastroenteritis can be managed with oral rehydration (Chapter 332).
It is important to address the underlying cause of the hypernatremia, if possible. The child with central diabetes insipidus should receive desmopressin acetate. Because this treatment reduces renal excretion of water, excessive intake of water must consequently be avoided to prevent both overly rapid correction of the hypernatremia and the development of hyponatremia. Over the long term, reduced sodium intake and the use of medications can somewhat ameliorate the water losses in nephrogenic diabetes insipidus (Chapter 524). The daily water intake of a child who is receiving tube feeding may need to be increased to compensate for high losses. The patient with significant ongoing losses, such as through diarrhea, may need supplemental water and electrolytes (Chapter 53). Sodium intake is reduced if it contributed to the hypernatremia.
Hyponatremia
Etiology and Pathophysiology
The causes of hyponatremia are listed in Table 52-2. Pseudohyponatremia is a laboratory artifact that is present when the plasma contains very high concentrations of protein (multiple myeloma, intravenous immunoglobulin infusion) or lipid (hypertriglyceridemia, hypercholesterolemia). It does not occur when a direct ion-selective electrode determines the sodium concentration in undiluted plasma, a technique that is used by the instruments used for measuring arterial blood gases. In true hyponatremia, the measured osmolality is low, whereas it is normal in pseudohyponatremia. Hyperosmolality, as may occur with hyperglycemia, causes a low serum sodium concentration because water moves down its osmotic gradient from the intracellular space into the extracellular space, diluting the sodium concentration. However, because the manifestations of hyponatremia are due to the low plasma osmolality, patients with hyponatremia resulting from hyperosmolality do not have symptoms of hyponatremia. When the etiology of the hyperosmolality resolves, such as hyperglycemia in diabetes mellitus, water moves back into the cells and the sodium concentration rises to its “true” value. Mannitol or sucrose, a component of intravenous immunoglobulin preparations, may cause hyponatremia due to hyperosmolality.
Table 52-2 CAUSES OF HYPONATREMIA
PSEUDOHYPONATREMIA
EXTRARENAL LOSSES
RENAL LOSSES
EUVOLEMIC HYPONATREMIA
HYPERVOLEMIC HYPONATREMIA
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
* Most cases of proximal renal tubular acidosis are not due to this primary genetic disorder. Proximal renal tubular acidosis is usually part of Fanconi syndrome, which has multiple etiologies.
Renal salt wasting occurs in hereditary kidney diseases, such as juvenile nephronophthisis and autosomal recessive polycystic kidney disease. Obstructive uropathy, most commonly a consequence of posterior urethral valves, produces salt wasting, but patients with the disease may also have hypernatremia as a result of impaired ability to concentrate urine and high water loss. Acquired tubulointerstitial nephritis, usually secondary to either medications or infections, may cause salt wasting, along with other evidence of tubular dysfunction. CNS injury may produce cerebral salt wasting, which is theoretically due to the production of a natriuretic peptide that causes renal salt wasting. In type II renal tubular acidosis (RTA), usually associated with Fanconi syndrome (Chapter 523), there is increased excretion of sodium and bicarbonate in the urine. Patients with Fanconi syndrome also have glycosuria, aminoaciduria, and hypophosphatemia due to renal phosphate wasting.
In SIADH, the secretion of ADH is not inhibited by either low serum osmolality or expanded intravascular volume (Chapter 553). The result is that the child with SIADH is unable to excrete water. This results in dilution of the serum sodium and hyponatremia. The expansion of the extracellular volume due to the retained water causes a mild increase in intravascular volume. The kidney increases sodium excretion in an effort to decrease intravascular volume to normal; thus, the patients has a mild decrease in body sodium. SIADH most commonly occurs with disorders of the CNS (infection, hemorrhage, trauma, tumor, thrombosis), but lung disease (infection, asthma, positive pressure ventilation) and malignant tumors (producing ADH) are other potential causes. A variety of medications may cause SIADH, including recreational use of 3,4-methylenedioxymethylamphetamine (MDMA, or “Ecstasy”), opiates, antiepileptic drugs (carbamazepine, oxcarbamazepine, valproate), tricyclic antidepressants, vincristine, cytoxan, and selective serotonin reuptake inhibitors. The diagnosis of SIADH is one of exclusion, because other causes of hyponatremia must be eliminated (Table 52-3). Because SIADH is a state of intravascular volume expansion, low serum uric acid and BUN levels are supportive of the diagnosis.
Table 52-3 DIAGNOSTIC CRITERIA FOR SYNDROME OF INAPPROPRIATE ANTIDIURETIC HORMONE SECRETION
Diagnosis
The history usually points to a likely etiology of the hyponatremia. Most patients with hyponatremia have a history of volume depletion. Diarrhea and diuretic use are very common causes of hyponatremia in children. A history of polyuria, perhaps with enuresis, and/or salt craving is present in children with primary kidney diseases or absence of aldosterone effect. Children may have signs or symptoms suggesting a diagnosis of hypothyroidism or adrenal insufficiency (Chapters 559 and 569). Brain injury raises the possibility of SIADH or cerebral salt wasting, with the caveat that SIADH is much more likely. Liver disease, nephrotic syndrome, renal failure, or congestive heart failure may be acute or chronic. The history should include a review of the patient’s intake, both intravenous and enteral, with careful attention to the amounts of water, sodium, and protein.
In patients with true hyponatremia, the next step in the diagnostic process is to clinically evaluate the volume status. Patients with hyponatremia can be hypovolemic, hypervolemic, or euvolemic. The diagnosis of volume depletion relies on the usual findings with dehydration (Chapter 54), although subtle volume depletion may not be clinically apparent. In a patient with subtle volume depletion, a fluid bolus results in a decrease in the urine osmolality and an increase in the serum sodium concentration. Children with hypervolemia are edematous on physical examination. They may have ascites, pulmonary edema, pleural effusion, or hypertension.
Hypovolemic hyponatremia can have renal or nonrenal causes. The urine sodium concentration is very useful in differentiating between renal and nonrenal causes. When the losses are nonrenal and the kidney is working properly, there is renal retention of sodium, a normal homeostatic response to volume depletion. Thus, the urinary sodium concentration is low, typically <10 mEq/L, although sodium conservation in neonates is less avid. When the kidney is the cause of the sodium loss, the urine sodium concentration is >20 mEq/L, reflecting the defect in renal sodium retention. The interpretation of the urine sodium level is challenging with diuretic therapy because it is high when diuretics are being used but low after the diuretic effect is gone. This becomes an issue only when diuretic use is surreptitious. The urine sodium concentration is not useful if a metabolic alkalosis is present; the urine chloride concentration must be used instead (Chapter 52.7).
Treatment
The child with hypovolemic hyponatremia has a deficiency in sodium and may have a deficiency in water. The cornerstone of therapy is to replace the sodium deficit and any water deficit that is present. The 1st step in treating any dehydrated patient is to restore the intravascular volume with isotonic saline. Ultimately, complete restoration of intravascular volume suppresses ADH production, thereby permitting excretion of the excess water. Chapter 54 discusses the management of hyponatremic dehydration.
Children with iatrogenic hyponatremia due to the administration of hypotonic intravenous fluids should receive 3% saline if they are symptomatic. Subsequent management is dictated by the patient’s volume status. The hypovolemic child should receive isotonic intravenous fluids. The child with nonphysiologic stimuli for ADH production should undergo fluid restriction. Prevention of this iatrogenic complication requires judicious use of intravenous fluids (Chapter 53).
Centers for Disease Control and Prevention (CDC). Application of lower sodium intake recommendations to adults—United States, 1999-2006. MMWR Morb Mortal Wkly Rep. 2009;58:281-283.
Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet. 2008;371:1624-1632.
Don M, Valerio G, Korppi M, et al. Hyponatremia in pediatric community-acquired pneumonia. Pediatr Nephrol. 2008;23:2247-2253.
Ellison DH, Berl T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064-2072.
Fenske W, Maier SKG, Blechschmidt A, et al. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123:652-657.
Karakas HM, Erdem G, Yakinci C. Osmotic demyelination syndrome in a 40-day-old infant. Diagn Interv Radiol. 2007;13:121-124.
Leung C, Chang WC, Yeh SJ. Hypernatremic dehydration due to concentrated infant formula: report of two cases. Pediatr Neonatol. 2009;50:70-73.
Linshaw MA. Back to basics: congenital nephrogenic diabetes insipidus. Pediatr Rev. 2007;28:372-380.
Modi N. Avoiding hypernatraemic dehydration in healthy term infants. Arch Child. 2007;92:474-475.
Rianthavorn P, Cain JP, Turman MA. Use of conivaptan to allow aggressive hydration to prevent tumor lysis syndrome in a pediatric patient with large-cell lymphoma and SIADH. Pediatr Nephrol. 2008;23:1367-1370.
Rivkees SA. Differentiating appropriate antidiuretic hormone secretion, inappropriate antidiuretic hormone secretion and cerebral salt wasting: the common, uncommon, and misnamed. Curr Opin Pediatr. 2008;20:448-452.
Verbalis JG, Goldsmith SR, Greenberg A, et al. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120:S1-S21.
52.4 Potassium
Potassium Metabolism
Body Content and Physiologic Function
The intracellular concentration of potassium, approximately 150 mEq/L, is much higher than the plasma concentration (see Web Fig. 52-3). The majority of body potassium is contained in muscle. As muscle mass increases, there is an increase in body potassium. There is thus an increase in body potassium during puberty, and it is more significant in males. The majority of extracellular potassium is in bone; <1% of total body potassium is in plasma.
mEq/L, is much higher than the plasma concentration (see Web Fig. 52-3). The majority of body potassium is contained in muscle. As muscle mass increases, there is an increase in body potassium. There is thus an increase in body potassium during puberty, and it is more significant in males. The majority of extracellular potassium is in bone; <1% of total body potassium is in plasma.
Hyperkalemia
Etiology and Pathophysiology
Three basic mechanisms cause hyperkalemia (Table 52-4). In the individual patient, the etiology is sometimes multifactorial.
Table 52-4 CAUSES OF HYPERKALEMIA
SPURIOUS LABORATORY VALUE
INCREASED INTAKE
TRANSCELLULAR SHIFTS
DECREASED EXCRETION
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
Normal doses of succinylcholine or β-blockers and fluoride or digitalis intoxication all cause a shift of potassium out of the intracellular compartment. Succinylcholine should not be used during anesthesia in patients at risk for hyperkalemia. β-Blockers prevent the normal cellular uptake of potassium mediated by binding of β-agonists to the β2-adrenergic receptors. Potassium release from muscle cells occurs during exercise, and levels can increase by 1-2 mEq/L with high activity. With an increased plasma osmolality, water moves from the intracellular space and potassium follows. This process occurs with hyperglycemia, although in nondiabetic patients, the resultant increase in insulin causes potassium to move intracellularly. In diabetic ketoacidosis, the absence of insulin causes potassium to leave the intracellular space, and the problem is compounded by the hyperosmolality. The effect of hyperosmolality causes a transcellular shift of potassium into the extracellular space after mannitol or hypertonic saline infusions. Malignant hyperthermia, which is triggered by some inhaled anesthetics, causes muscle release of potassium (Chapter 603.2). Hyperkalemic periodic paralysis is an autosomal dominant disorder caused by a mutated sodium channel. It results in episodic cellular release of potassium and attacks of paralysis (Chapter 603.1).
A wide range of primary adrenal disorders, both hereditary and acquired, can cause decreased production of aldosterone, with secondary hyperkalemia (Chapters 569 and 570). Patients with these disorders typically have metabolic acidosis and salt wasting with hyponatremia. Children with more subtle adrenal insufficiency may have electrolyte problems only during acute illnesses. The most common form of congenital adrenal hyperplasia, 21-hydroxylase deficiency, typically manifests in male infants as hyperkalemia, metabolic acidosis, hyponatremia, and volume depletion. Females with this disorder usually are diagnosed as newborns because of their ambiguous genitalia; treatment prevents the development of electrolyte problems.
Renin, via angiotensin II, stimulates aldosterone production. A deficiency in renin, a result of kidney damage, can lead to decreased aldosterone production. Hyporeninemia occurs in many kidney diseases, with some of the more common pediatric causes listed in Table 52-4. These patients typically have hyperkalemia and a metabolic acidosis, without hyponatremia. Some of these patients have impaired renal function, partially accounting for the hyperkalemia, but the impairment in potassium excretion is more extreme than expected for the degree of renal insufficiency.
A variety of renal tubular disorders impair renal excretion of potassium. Children with pseudohypoaldosteronism type 1 have hyperkalemia, metabolic acidosis, and salt wasting leading to hyponatremia and volume depletion; aldosterone values are elevated. In the autosomal recessive variant, there is a defect in the renal sodium channel that is normally activated by aldosterone. Patients with this variant have severe symptoms, beginning in infancy. Patients with the autosomal dominant form have a defect in the aldosterone receptor, and the disease is milder, often remitting in adulthood. Pseudohypoaldosteronism type 2, also called Gordon syndrome, is an autosomal dominant disorder characterized by hypertension due to salt retention and impaired excretion of potassium and acid, leading to hyperkalemia and metabolic acidosis. Activating mutations in either WNK1 or WNK4, both serine-threonine kinases located in the distal nephron, cause Gordon syndrome. In Bartter syndrome due to mutations in the potassium channel ROMK (type 2 Bartter syndrome), there can be transient hyperkalemia in neonates, but hypokalemia subsequently develops (Chapter 525).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


