Chapter 582 Disorders of Sex Development
Sexual Differentiation (Chapter 576)
In normal differentiation, the final form of all sexual structures is consistent with normal sex chromosomes (either XX or XY). A 46,XX complement of chromosomes as well as genetic factors such as DAX1 and the signaling molecule WNT-4 are necessary for the development of normal ovaries. Development of the male phenotype is even more complex. It requires a Y chromosome and, specifically, an intact SRY gene, which, in association with other genes such as SOX9, SF1, and WT1 and others (Chapter 576), directs the undifferentiated gonad to become a testis. Aberrant recombinations may result in X chromosomes carrying SRY, resulting in XX males, or Y chromosomes that have lost SRY, resulting in XY females.
Chromosomal aberrations may result in ambiguity of the external genitalia. Conditions of aberrant sex differentiation may also occur with the XX or XY genotype. The appropriate term for what was previously called intersex is disorders of sex development (DSD). This term defines a condition “in which development of chromosomal, gonadal or anatomical sex is atypical.” It is becoming more preferable to use the term “atypical genitalia” rather than “ambiguous genitalia.” Comparison with the previous terms and a new etiologic classification are seen in Tables 582-1 and 582-2. Some of the genes involved in disorders of sex development are listed in Table 576-1.
Table 582-1 REVISED NOMENCLATURE
| PREVIOUS | CURRENTLY ACCEPTED |
|---|---|
| Intersex | Disorders of sex development (DSD) |
| Male pseudohermaphrodite | 46,XY DSD |
| Undervirilization of an XY male | 46,XY DSD |
| Undermasculinization of an XY male | 46,XY DSD |
| 46,XY intersex | 46,XY DSD |
| Female pseudohermaphrodite | 46,XX DSD |
| Overvirilization of an XX female | 46,XX DSD |
| Masculinization of an XX female | 46,XX DSD |
| 46,XX intersex | 46,XX DSD |
| True hermaphrodite | Ovotesticular DSD |
| Gonadal intersex | Ovotesticular DSD |
| XX male or XX sex reversal | 46,XX testicular DSD |
| XY sex reversal | 46,XY complete gonadal dysgenesis |
From Lee PA, Houk CP, Ahmed SF, et al: Consensus statement on management of intersex disorders, Pediatrics 118:e488–e500, 2006.
Table 582-2 ETIOLOGIC CLASSIFICATION OF DISORDERS OF SEX DEVELOPMENT (DSD)
46,XX-DSD
Androgen Exposure
Disorder of Ovarian Development
Undetermined Origin
Associated with genitourinary and gastrointestinal tract defects
46,XY DSD
Defects in Testicular Development
Deficiency of Testicular Hormones
Defect in Androgen Action
Ovotesticular DSD
Sex Chromosome DSD
From Lee PA, Houk CP, Ahmed SF, et al: Consensus statement on management of intersex disorders, Pediatrics 118:e488–e500, 2006.
Development of the external genitalia begins with the potential to be either male or female (Fig. 582-1). Virilization of a female, the most common form of DSD, results in varying phenotypes (Fig. 582-2), which start from the basic genital appearances of the embryo (see Fig. 582-1).
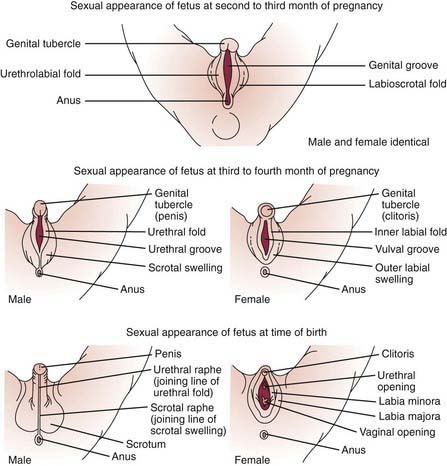
Figure 582-1 Schematic demonstration of differentiation of normal male and female genitalia during embryogenesis.
(From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 4, St Louis, 2002, Mosby, p 328.)
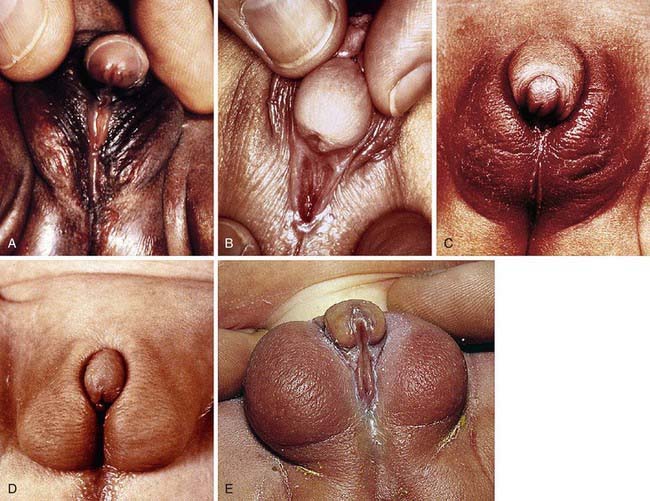
Diagnostic Approach to the Patient with Atypical or Ambiguous Genitalia
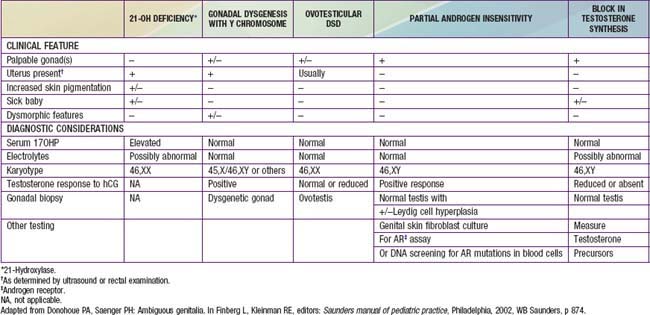
After a complete history and physical exam, the common diagnostic approach includes multiple steps, described in the following outline. These steps are usually performed simultaneously rather than waiting for results of 1 test prior to performing another, due to the sensitive and sometimes urgent nature of the condition. Careful attention to the presence of physical features other than the genitalia is crucial, to determine if a diagnosis of a particular multisystem syndrome is possible. These are described in more detail in Chapters 582.1, 582.2, and 582.3. A summary of many features of commonly encountered causes of DSD is provided in Table 582-3.
Diagnostic tests include the following:
582.1 46,XX DSD
Congenital Adrenal Hyperplasia (Chapter 570.1)
This is the most common cause of genital ambiguity and of 46,XX DSD. Females with the 21-hydroxylase and 11-hydroxylase defects are the most highly virilized, although minimal virilization also occurs with the type II 3β-hydroxysteroid dehydrogenase defect (see Fig. 582-1). Salt losers tend to have greater degrees of virilization than do non–salt-losing patients. Masculinization may be so intense that a complete penile urethra results, and the condition may mimic a male with bilateral cryptorchidism.
Aromatase Deficiency
In genotypic females, the rare condition of aromatase deficiency during fetal life leads to 46,XX DSD and results in hypergonadotropic hypogonadism at puberty because of ovarian failure to synthesize estrogen (see Fig. 568-1).
Bose HS, Pescovitz OH, Miller WL. Spontaneous feminization in a 46,XX female patient with congenital lipoid adrenal hyperplasia due to a homozygous frameshift mutation in the steroidogenic acute regulatory protein. J Clin Endocrinol Metab. 1997;82:1511-1515.
Diamond M, Sigmundson K. Sex reassignment at birth. Arch Pediatr Adolesc Med. 1997;151:298-304.
Diamond DA, Mitchell C, Lamb K, et al. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: attitudes and practices of pediatric urologists. J Pediatr. 2006;148:445-449.
Frade Costa EM, Bilharinho Mendonca B, Inacio M, et al. Management of ambiguous genitalia in pseudohermaphrodites: new perspectives on vaginal dilation. Fertil Steril. 1997;67:229-232.
Mendonca BB, Leite MV, DeCastro M, et al. Female pseudohermaphroditism caused by a novel homozygous mutation of the GR gene. J Clin Endocrinol Metab. 2002;87:1805-1809.
Moisan AM, Ricketts ML, Tardy V, et al. New insight into the molecular basis of 3β-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab. 1999;84:4410-4425.
Morishima A, Grumbach MM, Simpson ER, et al. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80:3689-3698.
Mullis PE, Yoshimura N, Kuhlmann B, et al. Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab. 1997;82:1739-1745.
Parisi MA, Ramsdell LA, Burns MW, et al. A gender assessment team: experience with 250 patients over a period of 25 years. Genet Med. 2007;9:348-357.
Saenger P. New developments in congenital lipoid adrenal hyperplasia and steroidogenic acute regulatory protein. Pediatr Clin North Am. 1997;44:397-421.
Wallien MS, Cohen-Kettenis PT. Psychosexual outcome of gender-dysphoric children. J Am Acad Child Adolesc Psychiatry. 2008;47:1413-1423.



