50 Disorders of Red Blood Cells
Etiology and Pathogenesis
Congenital Red Blood Cell Disorders
Thalassemias
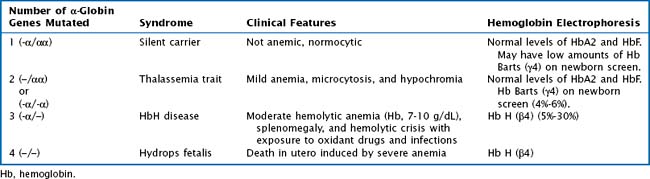
Whereas α -thalassemia is more commonly found in Southeast Asia, β-thalassemia is more common in Mediterranean countries. There are two α-globin genes located on chromosome 16. α-Thalassemias are usually the result of large gene deletions, causing a reduction in α-globin production. The severity of disease is directly related to the number of genes involved (Table 50-1).
Hemoglobinopathies
The hemoglobinopathies are a group of autosomal recessive inherited disorders characterized by synthesis of abnormal Hb molecules (i.e., S, and C). The most common and severe hemoglobinopathy is sickle cell disease, in which only HbS is produced. Chapter 53 of this book is dedicated to the in-depth discussion of sickle cell disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


