Fig. 14.1
Clinical suspicion for definite and non-criterial APS
14.2 Definite APS with Usual Clinical Manifestations
The clinical picture of APS is protean and very variable in the single patient, as thrombotic events can involve both arterial and venous vessels of any size and anatomical district and sometimes APS diagnosis requires a broad number of medical conditions to be ruled out (Table 14.1).
Table 14.1
Main differential diagnosis
SLE |
Behçet’s syndrome |
Demyelinating diseases (multiple sclerosis, Devic’s syndrome) |
Microangiopathic syndromes (TTP/HUS, HELLP)a |
Heparin-induced thrombocytopenia (HIT) |
Disseminated intravascular coagulation (DIC) |
Hyperviscosity syndromes |
Sneddon’s syndrome, Susac’s syndrome, Degos disease, Moyamoya syndrome (very rare) |
14.2.1 Systemic Lupus Erythematosus
A particular liaison exists between APS and SLE; the 2006 International consensus conference for the first time pointed out that the definition of secondary APS is not necessary and appropriate, since SLE and APS may represent a different spectrum of the same disease [1]. In clinical practice, we often challenge the following three scenarios:
1.
Patients who fulfill the international criteria for both SLE and APS. In this case, the two entities coexist.
2.
Patients with APS who also present some features of SLE, such as autoimmune hemolytic anemia, mild reduction of complement, and antinuclear antibodies (ANA) at borderline titer without any further specificity. These subjects can be considered as APS patients which likely will evolve into SLE in the future; in this case the early introduction of hydroxychloroquine therapy has been recommended [2].
3.
Patients with APS and renal involvement; in this case a significant titer of circulating ANA, the presence of other autoantibody specificities (such as antibodies anti-native DNA, anti-Sm, or anti-C1q), complement consumption, and above all the suggestive histological findings from renal biopsy are all crucial to distinguish SLE renal involvement from APS.
Whenever APS primarily involve the central nervous system (CNS), the main clinical challenge is the differential diagnosis with neuro-SLE, since both the conditions share similar neurological symptoms and MR findings [3]. The detection of a significant titer of specific circulating autoantibodies (e.g., anti-ribosomal P) and low complement levels should be suggestive of SLE. However, many patients with neuro-SLE show also circulating aPL; therefore, ischemic lesions seen on brain MR could be either the result of APS microthrombosis or alternatively of SLE inflammation. The discrimination between the two conditions could be very challenging for clinicians; however, this step is crucial since in the first case anticoagulants are the mainstay of therapy, while in the other SLE requires the use of high-dose steroids usually combined with immunosuppressants drugs [4].
14.2.2 Behçet’s Syndrome
Neuro-Behçet (NB) represents an important differential diagnosis for APS with predominant cerebral involvement. Of note both diseases can be responsible for cerebral venous sinus thrombosis and parenchymal lesions [5]. NB brain inflammatory lesions are typically located deeply at the level of basal ganglia and brainstem, while APS usually involves the subcortical and periventricular white matter areas. Differential diagnosis between the two conditions is crucial because in NB immunosuppressive treatment is required [6]. Moreover, a particular subset of Behçet patients is indeed very prone to vascular thrombosis that in these cases could represent the main clinical manifestation, further complicating the differential diagnosis with APS. Due to their primary inflammatory vascular origin, the thrombotic events in Behçet syndrome are usually better responsive to corticosteroid or immunosuppressive drugs than to anticoagulation [7]. Noteworthy the inappropriate use of anticoagulants in Behçet could be very dangerous when these drugs are used in patients with clinically occult pulmonary artery aneurisms, since the risk of rupture is very high in this setting [8]. Oral aphthosis is the most frequent clinical sign in Behçet; however, it lacks specificity due to its high frequency in the general and otherwise healthy population. Moreover, about 5 % percent of Behçet patients lack oral aphtosis. The same considerations are valid for HLA- B51 antigen, often wrongly used as a Behçet diagnostic biomarker, due to its high prevalence in Behçet patients, but aphtosis can also be found in approximately 30 % of unaffected people. HLA-B51 screening test and other laboratory investigations are not included in the classification criteria, since the diagnosis/classification of Behçet currently relies only on clinical findings [9]. Moreover, aPL could be present at low titer in Behçet patients. However, recurrent oral and genital (bipolar) aphthosis and uveitis when present are pathognomonic for Behcet’s disease and point to the correct diagnosis.
14.2.3 Demyelinating Diseases (Multiple Sclerosis and Neuromyelitis Optica)
Multiple sclerosis (MS) lesions can be hardly differentiated from neuro-APS on brain MR, since their radiologic morphology and location could be similar in the two conditions [10]. A further complication is represented by the not unusual finding of ANA and aPL positivity in MS patients, although often at very low titer. Lesions involving the periventricular and corpus callosum areas are more suggestive of SM than APS; moreover, the detection of oligoclonal bands from cephalospinal fluid further supports the diagnosis of demyelinating disease [11]. Neuromyelitis optica (NMO), previously named Devic’s syndrome, is a recently recognized disease, now considered a separate disorder from MS, since it recognizes a different inflammatory pathway. The presence of aquaporin-4 (anti-NMO) antibodies is considered the main pathogenetic player and a very useful diagnostic tool at the same time. The diagnosis of NMO should be suspected in patients who present with optic neuritis, usually more severe than in MS, and/or with longitudinally extended transverse myelitis, defined as a spinal inflammatory lesion involving at least three consecutive vertebrae. In this scenario testing patients for anti-NMO antibodies is strongly recommended [12]. NMO can coexist with SLE and APS in the same patient and this overlap created some degree of confusion in the past. Indeed, some neurological symptoms have been ascribed to SLE/APS rather than to NMO. This is the case, for example, of transverse myelitis frequently reported as an APS complication (among non-criteria clinical clues) but often with no data available on anti-NMO status. Recognizing NMO typical ocular/spinal pattern is hence considered fundamental for further investigating the presence of circulating anti-NMO, since this lead to the correct treatment with plasmapheresis or eventually with other immunosuppressive approach [13].
14.2.4 Microangiopathic Syndromes
Catastrophic antiphospholipid syndrome (CAPS), thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome all belong to the group of thrombotic microangiopathy syndromes; these disorders are histologically characterized by small vessel occlusion with hyaline thrombi and fibrin deposition, various degree of platelet consumption, intravascular hemolysis with red cells fragmentation producing schistocytes, and clinical/laboratoristic findings of organ dysfunction. Since aPL have been not infrequently detected in such conditions and since conversely APS does not uncommonly show some degree of microangiopathic involvement, the term MAPS (microangiopathic antiphospholipid syndrome) has been proposed as an “umbrella” to include many conditions [14, 15].
Thrombotic Thrombocytopenic Purpura
TTP, also named Moschowitz syndrome, is clinically characterized by severe neurological involvement, fever, thrombocytopenia, microangiopathic hemolytic anemia, and more rarely kidney dysfunction. Useful tools to diagnose this condition are the presence of schistocytes, elevated levels of circulating ultra large von Willebrand factor (ULVWF) multimers, and, in acquired autoimmune type, the detection of antibodies against the metalloproteinase-disintegrin ADAMTS13 [16]. Differential diagnosis could be hard since aPL have been occasionally reported in TTP patients; diagnostic clues helping in differential diagnosis are the rarity of severe episodes of thrombocytopenia in APS, while the occurrence of hemolytic anemia, the presence of schistocytes on peripheral blood smear, the elevated LDH levels, and the detection of ULVWF further support the diagnosis of TTP.
Hemolytic Uremic Syndrome
HUS is a disorder characterized by thrombocytopenia and microangiopathic anemia with microvascular thrombosis, tissue ischemia with necrosis, and renal failure. It has been distinguished into diarrhea-associated HUS, the most frequent and usually linked to Shiga-like toxin-producing bacteria (STEC-HUS), and diarrhea-nonassociated or atypical HUS. The latter constitutes only 5–10 % of HUS in children, but the majority of HUS in adults. Despite its rarity, atypical HUS is endowed with a very intriguing pathogenesis, since it can be triggered by genetical defect or by the presence of circulating proteins affecting the complement system [17, 18]. Clinical features suggesting HUS are the rapid onset of renal failure and the presence of microangiopathic anemia associated with thrombocytopenia; in the typical presentation the temporal occurrence of a previous episode of diarrhea as well as the absence of aPL further supports the diagnosis.
Hemolysis Elevated Liver Enzymes and Low Platelets
HELLP syndrome can complicate pregnancy, even in pregnant women with history of APS or aPL positivity. This condition is more common in multiparous women; 70 % of cases occur between the 27th and the 37th week of gestation, while in the remaining 30 % diagnosis is made during the postpartum period. The HELLP syndrome is characterized by upper right quadrant abdominal pain, nausea, vomiting, generalized edema, signs of hemolysis, elevated liver enzymes, severe thrombocytopenia, and renal dysfunction; noteworthy, severe hypertension is not usually found in HELLP syndrome, and this can help distinguish it from preeclampsia, even if the two conditions can coexist in 70–80 % of cases [19]. This scenario is even more complicated considering that some APS patients can experience HELLP syndrome during pregnancy, indicating that probably circulating aPL can affect the occurrence of HELLP in predisposed women [20].
For the main causes of recurrent miscarriage, see Fig. 14.2.
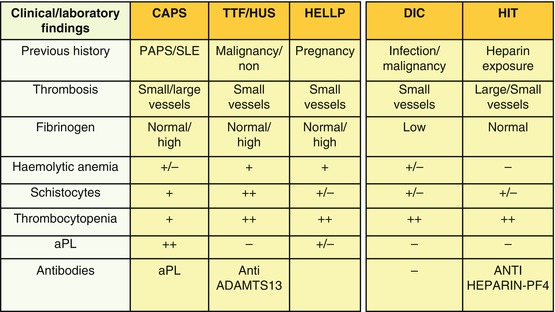
Fig. 14.2
Differential diagnosis between microangiopathies, DIC, and HIT
14.2.5 Heparin-Induced Thrombocytopenia
Another condition that must be considered in the differential diagnosis of APS is heparin-induced thrombocytopenia (HIT), clinically characterized by thrombocytopenia and thrombotic events and histologically by multiple thrombotic occlusions of small vessels [21]. This entity should be suspected early during heparin treatment and always considered as potentially fatal. Two forms of HIT have been described: type I HIT, not immune-mediated, has a favorable prognosis since it is not associated with thrombotic events; type II HIT, with an underlying immunological mechanism, is associated with microthrombosis involving multiple anatomical sites. Type I HIT occurs in approximately 10 % of patients undergoing both unfractionated heparin (UH) or low molecular weight heparin (LMWH) treatment and is thought to be caused by direct platelet activation from heparin. It occurs very early usually within the first 4/5 days of therapy and resolves without treatment and without any complication. Type II HIT is conversely a dreaded complication, potentially involving 1–5 % of patients undergoing heparin therapy. Approximately 50 % of patients with type II HIT may experience thrombotic events affecting both the arterial and the venous vascular district. The risk of mortality in such cases is estimated to be about 20–30 %. The likelihood of developing type II HIT is considered to be between 10 and 40 times higher when unfractionated heparin is used compared to LMWH. It should be suspected when the platelet count falls by about 50 % compared to baseline after 5–15 days from the beginning of heparin therapy. Type II HIT can present in three ways: (a) production of specific antiplatelet antibodies without thrombocytopenia; (b) antibodies associated with thrombocytopenia but not with thrombotic complications; and (c) antibodies, thrombocytopenia, and thrombosis. The pathogenesis of HIT has been recently better defined and the complex heparin-platelet factor 4 (PF4) identified as the major immunological target of antibody response. During the early phase heparin binds to PF4, generating the heparin-PF4 complex toward which IgG antibodies are produced. In HIT the heparin-PF4-IgG complex is also able to activate platelets via FcγRIIa receptor, causing microthrombosis and thrombocytopenia. The resulting platelet activation is also associated with serotonin release and thrombin generation. HIT laboratory diagnosis, based on detection of antibodies to heparin-PF4 complex, can be made mainly using the following two methods: a serotonin release assay (SRA), characterized by low sensitivity but very specific for HIT, or alternatively an ELISA test, more sensitive than the previous one, but less specific for HIT; neither of the above cited tests anyways represents the gold standard and the diagnosis is currently based on probability according to a compatible clinical scenario [22].
Finally HIT management is very problematic and consists of both the immediate discontinuation of heparin and the beginning of an alternative anticoagulation strategy with (a) direct thrombin inhibitors, (b) heparinoids, and (c) pentasaccharides. The differential diagnosis with APS can be difficult because both can be associated with low platelet count and thrombosis, even if thrombocytopenia is generally more severe in HIT than in APS. Obviously previous heparin exposure, the precise timing of thrombocytopenia insorgence, and the absence of circulating aPL are all points to make a correct diagnosis.
14.2.6 Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is a serious disorder, secondary to infections, surgery, chronic inflammatory diseases, different obstetrical diseases, and solid and hematological malignancies. It is characterized by disseminated microvascular thrombosis, consumptive coagulopathy, and hemorrhagic diathesis, which can eventually lead to multiple organ failure. Laboratory features of DIC are thrombocytopenia, prolonged clotting times, reduced plasma fibrinogen, and increased levels of fibrin degradation products. Interestingly, DIC could both mimic and complicate APS, especially in its catastrophic form; indeed, CAPS is a condition with widespread intravascular thrombosis resulting in multiorgan failure. More frequently DIC presents thrombotic and hemorrhagic complications at the same time [23]
For differential diagnosis between microangiopathies, HIT, and DIC, see Fig. 14.3.
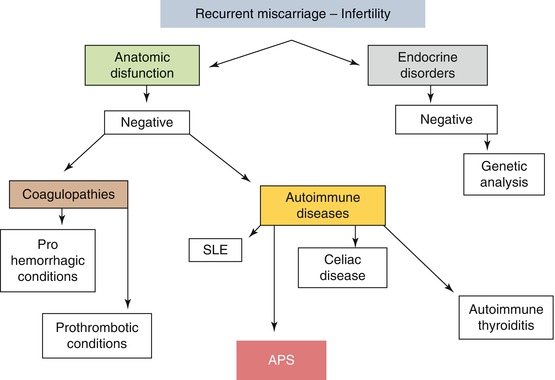
Fig. 14.3
Main causes of recurrent miscarriage
14.2.7 Hyperviscosity Syndromes
Patients with hyperviscosity syndrome (e.g., in multiple myeloma or Waldenstrom’s macroglobulinemia) may present cerebral ischemia and transitory ocular problems, such as amaurosis fugax, and sometimes a differential diagnosis with APS may be needed.
14.2.8 Other Clinical Clues
The malignant atrophic papulosis (MAP), also known as Köhlmeier-Degos disease, needs to be differentiated from APS. It exists in two variants: the “benign” type with only cutaneous involvement and the “malignant” type with internal organ involvement in addition to the typical cutaneous lesions. These latter systemic type can indeed involve not only the skin but also the gastrointestinal tract or other critical anatomical districts such as eye, heart, liver, kidneys, and CNS. The differential diagnosis is especially with neuro-APS or CAPS. Typical skin lesions are erythematous papules that evolve into atrophic scars with a porcelain white center, histopathologically indistinguishable from certain cutaneous lesions of SLE. Gastrointestinal involvement is present in approximately 60 % of patients and is the most common cause of death, while the CNS involvement is characterized by single or multiple infarcts in the brain and spinal cord. The differential diagnosis is based on (a) the presence of typical skin lesions, (b) gastrointestinal symptoms, and (c) the absence of aPL [24].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


