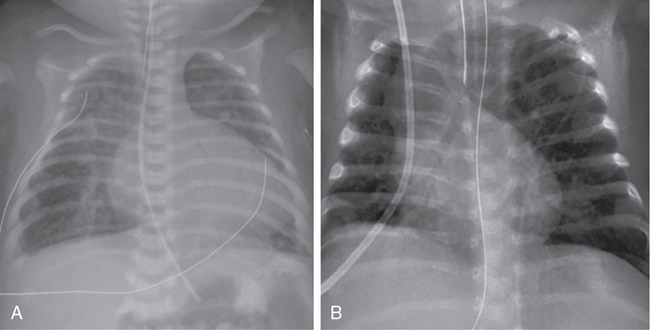
Shazia Bhombal, Ganga Krishnamurthy, Jay D. Pruetz, Yogen Singh The incidence of congenital heart disease (CHD) is estimated at approximately 8 per 1000 live births,1 with up to 25–30% of CHD deemed critical congenital heart disease (CCHD). CCHD includes lesions that necessitate early interventions to avoid significant morbidity and mortality and require multidisciplinary subspecialty care from pediatric cardiologists, cardiothoracic surgeons, neonatologists, anesthesiologists, and pediatric cardiac intensivists. Detection of CCHD by prenatal ultrasound (fetal anomaly screening) or by newborn pulse oximetry screening can occur prior to decompensation resulting in decreased morbidity and mortality in this vulnerable population.2–4 Although there have been remarkable advances in ultrasound technology and an increasing prenatal detection rate, a significant proportion of cases of CCHD are diagnosed postnatally.5 In the United States prenatal detection of CCHD has gradually increased from 26% in 2006 to 43% in 2012, with improvements in ultrasound technology and improved screening guidelines that emphasized the outflow tract views.6 As such, CCHD may be unknown at birth, and neonatologists or non-cardiologist acute care physicians may be the “first call” for a decompensating infant with CCHD. Congenital heart disease remains the leading cause of infant mortality at approximately 40% of all deaths resulting from birth defects; thus familiarity with signs and symptoms of CCHD is paramount in the care of a decompensating neonate.7 This chapter focuses on understanding the presenting pathophysiology and outlines the initial management by neonatal providers for a patient with CCHD. Critical congenital heart disease traditionally refers to lesions deemed necessitating surgical or catheter-based procedures in the first year of life,8 with the focus of this chapter on those requiring life-sustaining intervention within the first postnatal month. Neonates with CCHD can manifest with varying degrees of severity due to several different pathophysiologic states: (1) increasing cyanosis due to lack of mixing of systemic and pulmonary venous return or inadequate pulmonary arterial blood flow; (2) signs of hypoperfusion resulting from inadequate systemic blood flow due to obstruction of systemic blood flow or decreased systolic function; and (3) respiratory distress due to excessive pulmonary flow or pulmonary venous obstruction. Neonates may have features of more than one presentation type depending on the lesion type and postnatal age. Timing of presentation will depend on type of CHDs ([i] ductal-dependent lesions and timing of PDA closure, e.g., hypoplastic left heart syndrome [HLHS], [ii] patent foramen ovale [PFO]/central mixing dependent, e.g., transposition of the great arteries [TGA], and [iii] obstructed pulmonary venous return), the rate of drop in pulmonary vascular resistance (PVR), and associated heart lesions (see Table 30.1). In a study assessing time to significant hemodynamic compromise in the setting of undiagnosed CCHD, defined as severe metabolic acidosis, cardiac arrest, lab evidence of renal or hepatic insult, or seizure, most (83%) occurred after 12 postnatal hours. Aortic arch obstructive lesions comprised approximately 90% of cases with significant hemodynamic compromise that were potentially preventable.9 In addition, early diagnosis of CCHD (antenatal or pre-discharge) had a higher survival advantage versus late diagnosis following discharge from hospital (16% vs. 27%).10 Adapted from Desai et al.69 *Corresponds to respiratory compromise with TOF with absent pulmonary valve in column. CAVC, complete atrioventricular canal CCHD, critical congenital heart disease; CHF, congestive heart failure; HLHS, hypoplastic left heart syndrome; PDA, patent ductus arteriosus; PS, pulmonary stenosis; PVR, pulmonary vascular resistance; TAPVC, total anomalous pulmonary venous connection; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; VSD, ventricular septal defect. Within CCHD, five categories of lesions prevail, with recognition that many lesions can fall into more than one category: (1) ductal-dependent pulmonary blood flow, (2) ductal-dependent systemic blood flow, (3) pulmonary venous obstruction, (4) mixing lesions, and (5) transposition of the great arteries (see Table 30.2). CCHD lesion types with inadequate aortic or pulmonary outflow are termed “ductal dependent”, given the need for flow through the ductus arteriosus to provide or augment either pulmonary or systemic blood flow. *Pulmonary flow may be supplied by multiple aortopulmonary collaterals (MAPCAs) and not be DD. +Most commonly normally related great vessels, with DD pulmonary flow; can be transposed great vessels and reliant on ductus to augment systemic flow. In patients with ductal-dependent pulmonary circulation, prostaglandin E1/E2 is required to augment pulmonary blood flow. This obstruction can occur anywhere along the path through the right side of the heart, from the tricuspid valve (e.g., tricuspid atresia with normally related great vessels) to the right ventricular outflow tract (RVOT, e.g., pulmonary atresia with intact septum, and tetralogy of Fallot with severe pulmonary stenosis or Ebstein’s anomaly with pulmonary obstruction) (see Figure 30.1). Flow through the ductus arteriosus in patients with ductal-depending pulmonary circulation will traverse from the aorta to the lungs, with increasing pulmonary flow (Qp) over time as the PVR decreases. In patients with ductal-dependent pulmonary circulation, decreased saturations could be a result of decreased Qp if pulmonary flow decreases, such as in the case of ductal closure or restriction, or due to decreased pulmonary venous saturation related to pulmonary edema in cases with increased Qp as PVR decreases. Chest radiograph can help differentiate between these conditions with oligemic lung fields as pulmonary flow decreases and plethoric lung fields in patients with increased Qp (see Figure 30.2). Lesions with ductal-dependent systemic flow include left-sided obstructive lesions that limit systemic blood flow without the presence of a patent ductus arteriosus (PDA), such as interrupted aortic arch, severe coarctation, and hypoplastic left heart syndrome (HLHS) (see Table 30.1). As the ductus arteriosus (DA) restricts, patients may present in cardiogenic shock with hypoperfusion and acidosis and in extreme cases with death when the PDA closes. As the DA starts to constrict, pulses and perfusion can diminish, and if it continues to close completely, the patient may present in extremis with shock. Even once the DA is reopened successfully, the cardiac function may need time to recover as a result of the acidosis and the increased afterload on the left ventricle prior to surgical intervention. Early intervention may be necessary in patients with ductal-dependent systemic circulation. While most cases of HLHS can be stabilized by maintaining DA patency to provide adequate systemic flow, approximately 6–20% of HLHS cases have an intact or restrictive atrial septum, with pulmonary venous obstruction.11–13 These infants present with early decompensation after birth, similar to d-TGA with restrictive atrial septum/intact atrial septum (RAS/IAS) though related to different pathophysiologic mechanism related to lack of egress from the left atrium and pulmonary vasculopathy. Infants with a restrictive or intact atrial septum present a time-critical emergency as they require immediate intervention for the creation of mixing at the atrial level despite successfully keeping the DA patent. Catheter-based or surgical intervention on the atrial septum is imperative to ensure adequate atrial mixing; nevertheless, even with intervention, mortality is still high.13 Catheter-based fetal septal interventions have been attempted in these infants, and while a recent analysis demonstrated procedural successes, this has not yet translated into improved survival when compared with a cohort of HLHS with patent atrial septum.14 Not all critical CHDs are necessarily ductal dependent, yet many still require intervention in the first postnatal month. CCHD with obstructed venous flow such as obstructive total anomalous pulmonary venous connection (TAPVC), mixing lesions such as truncus arteriosus, and transposition of the great arteries (TGA) are not ductal-dependent lesions by definition; however, patients with TGA may have some benefit in use of PGE while obstructive TAPVC may either improve transiently or even worsen with its use. With TGA, the blood moves in parallel circuits, while not a traditional mixing lesion; specifically, systemic flow returns to the right heart and then is ejected out the aorta back to the body, while the pulmonary venous flow returns to the left heart and is ejected out the pulmonary artery back to the lungs. An atrial septal defect is required to allow oxygenated blood from the left atrium to the right atrium and out the aorta to the body (see Figure 30.3). In patients with desaturation utilization of PGE will allow increased pulmonary flow to enter the left atrium and encourage further mixing at the atrial level. A common misconception is that utilization of PGE providing ductal patency provides mixing at the ductus level. In fact, what the DA often provides is effective pulmonary blood flow, such that deoxygenated systemic blood can get to the lungs and then return to the left atrium oxygenated and mix at the atrial level. It is important to remember that in patients with TGA, the expectation is for flow to traverse from the aorta with higher systemic vascular resistance across the PDA to the pulmonary arteries and lung as PVR falls. In some patients with a dysregulated postnatal transition, elevated PVR may further limit pulmonary blood flow and the efficacy of oxygenation, necessitating transient use of pulmonary vasodilators. A ventricular septal defect alone may not allow for adequate mixing as flow across the VSD is also dependent on the relative downstream resistances. Another physiologic consideration is the potential for reversed differential saturation in patients with d-TGA and pulmonary hypertension, with preductal saturations lower than post-ductal saturations. In these patients pulmonary venous flow enters the left atrium, into the left ventricle, and out the RVOT. In patients with elevated PVR in comparison to systemic vascular resistance (SVR), oxygenated blood will shunt right to left across the PDA and down the descending aorta, while systemic flow will enter the right atrium, with some mixing of oxygenated blood from the left to right atrium into the right ventricle, out the aorta. Both patients with obstructive TAPVC and TGA with restrictive/intact atrial septum will present at or soon after birth with hypoxemia (decreased saturation) and will deteriorate regardless of ductal flow. In patients with obstructive TAPVC, which is most commonly infradiaphragmatic, PGE initiation may be detrimental as it may increase flow into a vascular bed with a downstream obstruction. In these cases, what is needed to prevent further morbidity and mortality is a rapid and definitive surgical repair to reconnect the pulmonary veins to the left atrium. TAPVC is particularly challenging to diagnose prenatally15 and thus should be on the differential of a neonate with respiratory distress after delivery and similar CXR findings to meconium aspiration. Finally, patients with truncus arteriosus have no protection of their pulmonary vascular bed, with only one valve separating intracardiac flow to systemic and pulmonary flow, with high risk of development of pulmonary vascular disease.16 As PVR drops, increasing flow will steal from the systemic circulation to the pulmonary circulation, placing neonates with truncus arteriosus at high risk of end-organ hypoperfusion. In particular, these patients have a higher risk of developing necrotizing enterocolitis (NEC) from hypoperfusion to the gut.17 Although there are some situations where initiation of PGE may worsen a patient’s condition, in most cases of CCHD (including TAPVC), PGE will either help or not significantly harm outcomes. Thus, from a neonatologist’s perspective, it is always reasonable to start prostaglandin when critical CHD is suspected until underlying anatomy is delineated by a pediatric cardiologist. Cyanotic heart diseases, with hypoxemia as the primary presenting symptom, result from mixing of systemic flow (Qs) and pulmonary flow (Qp). Cyanosis occurs due to two mechanisms: (1) intracardiac shunting of deoxygenated blood from the right side of the heart to the left-side systemic circulation secondary to cyanotic CHD and (2) extrapulmonary right-to-left shunts at the levels of the PDA or PFO in acute pulmonary hypertension (PH). In the former, related to shunting of intracardiac mixing, the blood leaving the heart is already mixed, with both pre- and post-ductal saturations reflecting hypoxemia. The etiology includes both right-sided obstructive lesions (e.g., tricuspid atresia, tetralogy of Fallot, pulmonary atresia) and left-sided obstructed lesions (e.g., hypoplastic left heart syndrome), and mixing lesions without obstruction (e.g., truncus arteriosus, total anomalous pulmonary venous return, transposition of the great arteries). These seven cyanotic lesions with intracardiac shunting are the target of the postnatal CCHD pulse oximetry screen (see Table 30.3). Cyanosis due to extrapulmonary shunts for CCHD relates to cases of arch obstruction such as interrupted aortic arch with a PDA. In this situation measurement of preductal saturation (right arm) and post-ductal saturation (right leg) may show a significant difference between pre- and post-ductal PaO2, indicating that the blood is shunted at the level of PDA from pulmonary to systemic circulation. Therefore it is important for neonatologists to recognize that an exclusive pre-/post-ductal saturation gradient is not pathognomonic of acute PH. Adapted from Oster et al.70 While hypoxemia is present in most neonates with CCHD, appreciation of cyanosis on clinical exam is challenging, with 4–5 g of deoxygenated Hb required to produce visible cyanosis. In a neonate with Hb 20 g/dL cyanosis would be noted clinically at an oxygen saturation of <80%, but at less than 60% in a neonate with a Hb of 10 g/dL8. Of note, neonates with CCHD and cyanosis often appear comfortable without respiratory distress, as opposed to patients with cyanosis and respiratory compromise leading to poor gas exchange and/or pulmonary hypertension with right to left ductal shunting. With recognition that early diagnosis of CCHD improves outcomes, pulse oximetry screening was added to the US-recommended Uniform Screening Panel for newborns in 2011.18 In a state review the estimated numbers of CCHD were 0.17% of the population (approximately 136 patients per year), 48 with a critical left heart lesion and 7 with missed critical left heart lesions. These numbers are more impressive when compared to the routine statewide screening program, which picked up 26 cases of galactosemia, 5 of phenylketonuria, and 0 of maple syrup urine disease. This reflects the importance of a screening program for critical CHD.19 Prior to implementation of universal pulse oximetry screening, a state registry noted that CCHD was not diagnosed during initial hospitalization in approximately 23% of patients with CCHD. Patients born in level 1 or level 2 hospitals had later detection of CCHD than those born in a level 3 hospital, indicating that pulse oximetry screening may be particularly useful in level 1 and level 2 units.20 However, while addition of the pulse oximetry screening to the existing screening tools will help in detecting up to 80–92% of CCHD, some lesions remain under-detected, particularly left-sided heart obstruction. (Table 30.3).9 Hence a neonatologist who is often the first point of contact for seeing these infants should have a low threshold for suspecting underlying CHD and initiating therapy such as prostaglandin E1 (PGE) and obtaining an echocardiogram. Echocardiography is the investigation of choice to diagnose CHD at bedside; however, pediatric cardiology services may not be readily available in all centers. Institutions with neonatal hemodynamic consult services where a neonatologist trained in targeted neonatal echocardiography or neonatologist-performed echocardiography (TNE or NPE, respectively) can utilize imaging to differentiate between varying pathologies. NPE-trained personnel have demonstrated the ability to identify structural heart disease when echocardiography was obtained prior to imaging by the cardiac service.21 While the goal of NPE is not to diagnose congenital heart disease, in a decompensating patient, it may be life-saving and provide earlier access to pediatric cardiology in centers where these services are not routinely available. Therefore all first echocardiograms must be comprehensive and, if not performed by a pediatric cardiologist, include all the necessary views/sweeps to enable exclusion of major forms of congenital heart disease. These images may be remotely reviewed by a pediatric cardiologist with imaging expertise. However, it is important to recognize that imaging may not be readily available for diagnosis of CHD and understanding the physiology and timing of presentation is integral to ensuring that CCHD is considered and managed in a neonate. The initial presenting symptoms to a neonatologist may include limitation to systemic blood flow. Hypoperfusion may be observed in all categories of CCHD lesions. Hypoperfusion resulting from decreased systemic blood flow can manifest with lactic acidosis, end-organ dysfunction, cool extremities, and in severe cases circulatory collapse with shock. In a patient with ductal-dependent pulmonary blood flow (e.g., pulmonary atresia with intact ventricular septum), as PVR drops in the presence of a large PDA, increased flow left to right from the arch to the pulmonary vasculature can lead to decreased systemic flow, resulting in metabolic acidosis. This can be more pronounced in patients with mixing lesions without obstruction, such as truncus arteriosus. As PVR falls, increased ductal flow is shunted to the pulmonary vasculature unimpeded through systole and diastole, with patients with truncus arteriosus noted to have higher risk for development of NEC.17 Finally, patients with CCHD with ductal-dependent systemic flow are at risk of systemic hypoperfusion; in particular, patients with HLHS are at higher risk for development of NEC compared with other congenital heart disease.17,22 Transition at birth from fetal to postnatal circulation involves lung expansion and a drop in PVR with loss of the low-resistance placental circulation, resulting in increased systemic vascular resistance, diminishing contribution of fetal intracardiac shunts, and a shift to reliance on oxygen from the lungs rather than the placenta.23,24 Most patients with CCHD remain relatively asymptomatic during the postnatal transition aside from lower oxygen saturation if a pulse oximeter is placed. Timing of presentation depends on the type of lesion and potential additional factors (Table 30.1). Lesions with severe obstruction to flow, limitation of cardiac output, or respiratory compromise resulting from CCHD present immediately after birth. CCHD with instability during the immediate postnatal transition can be grouped into four categories: (1) inadequate pulmonary venous egress, such as in obstructed TAPVC and subtypes HLHS with intact or restrictive atrial septum; (2) inadequate intracardiac mixing of oxygenated blood to the systemic circulation in some cases of d-transposition of the great arteries (d-TGA) with restrictive atrial shunt; (3) associated airway anomalies which compromise the ability to ventilate such as severe cases of Ebstein’s anomaly and tetralogy of Fallot (TOF) with absent pulmonary valve (APV); and (4) inadequate cardiac output in cases of severe fetal arrhythmias and/or decreased cardiac function.25 Additionally, identification of the fetus with CCHD and hydrops may allow time for optimal delivery planning, weighing risks of early delivery with worsening hydrops. Patients with certain CCHD that can lead to an increase in central venous pressure may be at risk for fetal hydrops, such as lesions with significant atrioventricular (AV) valve regurgitation (e.g., AV septal defects, Ebstein’s anomaly, tricuspid valve dysplasia). Two factors may contribute to development of hydrops: (1) the fetal myocardium is less compliant and does not tolerate small changes in preload well (due to diastolic stiffness) and (2) fetuses have increased capillary permeability, lower osmotic pressure, and reliance on lymphatics for drainage, contributing to increased extracellular fluid volume with small increases in CVP26,27 Prenatal diagnosis and fetal hemodynamic monitoring of these critical forms of CHDs can allow for multidisciplinary planning for delivery and well-coordinated care during transition, as well as providing prenatal counseling to families. Prenatal diagnosis of CCHD allows for adequate time to counsel families, create a perinatal transition plan, and prepare the multidisciplinary team for a well-coordinated delivery with postnatal care.28 Delivery and transition plans must take into account the underlying cardiac anatomy, anticipated physiologic changes during transition from fetal to postnatal life, rate of risk of deterioration, the need for emergent neonatal intervention, and potential definitive surgical options. Risk stratification systems are designed according to the severity level of the CHD and include associated neonatal action plans. These stratification systems can designate the need for PGE, the mode of delivery (MOD), appropriate level of perinatal and neonatal care services available, and proximity to immediate access to cardiology and cardiothoracic surgery care. Risk stratification and management systems using level of care (LOC) for neonates prenatally diagnosed with CHD have been shown to be highly accurate at predicting the postnatal care and need for emergent intervention at birth.29,30 The four levels of care, with level 1 being lowest and level 4 being highest risk for compromise, are denoted based on the type of cardiac lesion and agreed on by the perinatal team, including obstetrics, neonatology, CT surgery, and cardiology, helping to guide the delivery and transition plan.25,30 These classification strategies have been highly reproducible, with the exception of d-TGA, due to the difficulty in determining the risk for postnatal atrial level restriction. As a result, all d-TGA cases are automatically upgraded from LOC 3 to LOC 4 status.30 LOC 1 patients are low risk, not expected to necessitate support or intervention in the immediate newborn period, and include patients with atrial septal defects and ventricular septal defects. Level 2 denotes intermediate risk including patients with severe systemic or pulmonary outflow tract obstruction with ductal dependent flow. Patients with lesions in this category, such as HLHS, are typically stable in DR but require prostagladin for ductal patency and transfer to cardiac center for intervention. Level 3, identifies patients at high risk for immediate postnatal catheter intervention or surgery such as HLHS with restrictive atrial septum or CHD with arrhythmia or decreased function. Finally, level 4 care indicates a high-risk population requiring immediate intervention postnatally, including HLHS with intact atrial septum, d-TGA with RAS/IAS, and severe Ebstein’s with hydrops or incessant arrhythmia. Level 3 and 4 deliveries via planned cesarean section versus induction near term must be discussed with the multidisciplinary team to assess ability for coordination of care and at a center with interventional/surgical services for rapid access.25,30 If unable to deliver at a cardiac center and concern for rapid decompensation, particularly level 4 patients, transport team should be ready to transfer to a center with cardiac intervention services as soon as feasible (Table 30.4). Another institution implemented a slight variation to the LOC 1–4 risk stratification, separating CHD by need for emergent neonatal cardiac intervention (ENCI). In their model the classification system correctly stratified 90.4% of prenatally diagnosed CHD patients based on intervention need. No neonate with ENCI level 1 (low risk, e.g., ASD, VSD, mild PS) or level 2 (medium risk, not PGE dependent, e.g., complete AV canal, TOF/PS, truncus) needed neonatal emergent intervention.29
Chapter 30: Critical congenital heart disease: What should the neonatologist know?
Introduction
Clinical presentation of CCHD
Soon After Birth (Severe Cyanosis With Inadequate Mixing or Respiratory Compromise*)
Within First Days to 1–2 Weeks (PDA Dependent Perfusion)
Within First Month (PVR Decreases, CHF, Cyanosis, Hypoperfusion)
Physiology of CCHD
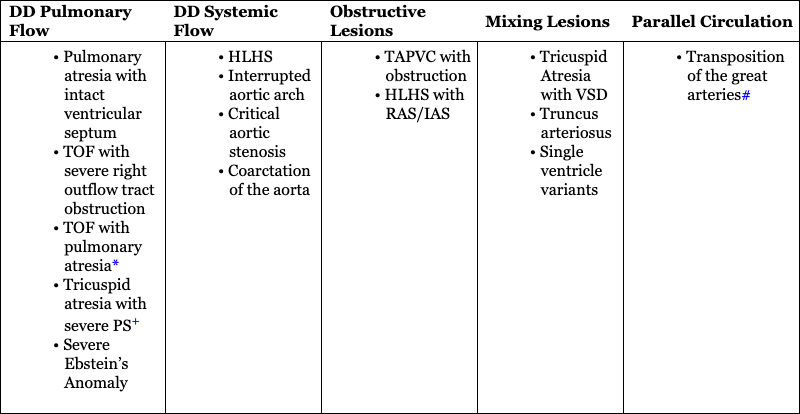
DD Pulmonary Flow
DD Systemic Flow
Obstructive Lesions
Mixing Lesions
Parallel Circulation
Ductal-dependent pulmonary circulation
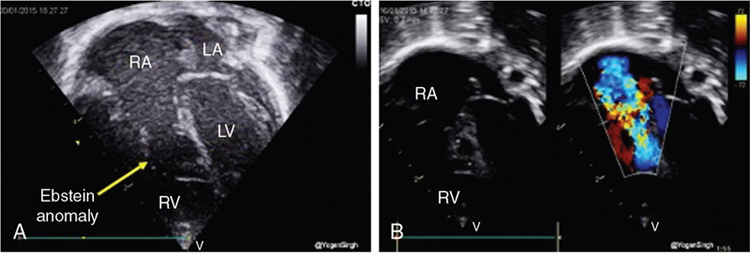
Ductal-dependent systemic circulation
Non-ductal-dependent CCHD
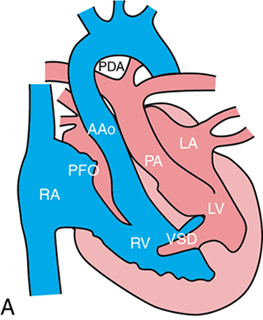
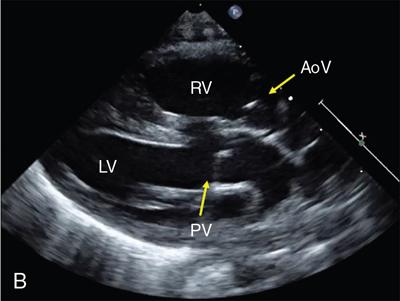
Clinical presentation of CCHD
Cyanosis and postnatal pulse oximetry screening
Primary Targets of CCHD Screen
Secondary Targets of CCHD Screen
Hypoperfusion
Postnatal transition and instability with CCHD
Perinatal management strategies to optimize postnatal transition
Critical congenital heart disease: What should the neonatologist know?
Targets of the CCHD Screen, 7 Primary Targets, 5 Secondary Targets Typically Associated With Some Hypoxemia70



