Fig. 13.1
Pathology of craniopharyngioma. a Adamantinomatous craniopharyngioma characterized by trabecules of stratified squamous epithelium with peripheral palisading of basaloid cells (black arrows). A microcystic component (on the right) contains degenerated squamous cells (*), keratin debris, and microcalcifications (black arrowheads). A solid component (on the bottom right), consists of foreign body multinucleated giant cells (white arrowheads) reactive to keratin debris and squames (white arrows). The lesion’s periphery consists of dense fibrous tissue. b Another component of craniopharyngioma with extensive fibrosis, cholesterol clefts, hemosiderin deposition, multinucleated giant cell reaction (arrows) and squames (arrowheads)
Greater than 90 % of all pediatric craniopharyngiomas are adamantinomatous, with fewer than 2 % pure papillary and the remainder exhibiting mixed features [10–13].
Molecular/Genetic Pathology
Disruptions in apoptotic pathways involving beta-catenin and Wnt may contribute to neoplastic transformation of craniopharyngiomas [8].
Growth factors and angiogenic peptides have been implicated in craniopharyngioma growth and development [8]. These are relevant not only for the light that they shed on the basic mechanisms of tumorigenesis but also because they facilitate development of novel methods of tumor detection and follow-up, such as recent data suggesting that urine may be able to noninvasively detect brain tumors, specifically including craniopharyngiomas [14, 15].
Incidence and Prevalence
In the United States, approximately 340 craniopharyngiomas are diagnosed annually in the combined pediatric and adult population, of which 100 are in children between 0 and 14 years of age [16].
Craniopharyngioma has an incidence of 1.3 cases diagnosed per million person-years [16].
Age Distribution
Sex Predilection
There is no sex predilection for craniopharyngioma.
Geographic Distribution
None.
Risk Factors—Environmental, Life Style
None.
Relationships to Other Disease States, Syndromes
Craniopharyngiomas represent 5–10 % of all intracranial tumors in children in many series and represent more than half of all sellar region tumors in this population [11, 16, 17].
They are the most common nonglial intracranial tumors in children [18].
Presentation
Symptoms
The most common clinical findings include sequelae of increased intracranial pressure (ICP) due to mass effect and hydrocephalus (especially headache, nausea and vomiting), visual loss and/or endocrinologic dysfunction [6, 18, 19]. These signs and symptoms are directly related to the location of the tumor and variations in the site of origin help to explain variations in presentation. Generally, cranipharyngiomas exhibit one of three growth patterns—prechiasmatic (affecting the optic nerves/chiasm), retrochiasmatic (affecting the hypothalamus, optic tracts, and drainage of cerebrospinal fluid (CSF)), and sellar (affecting hormonal function).
Vision
Endocrinologic Dysfunction
Similarly insidious is the development of endocrinologic dysfunction, which may remain unnoticed for long periods of time due to the often subtle onset of symptoms, such as growth delay. While deficiency of growth hormone is the most common endocrinopathy found in the setting of craniopharyngioma (followed by hypothyroidism and diabetes insipidus), abnormalities of any and all of the pituitary hormones may be manifest and careful retrospective analysis of patients reveals some form of endocrine dysfunction in 60–90 % of patients at diagnosis [18, 21].
Hypothalamic Injury
Hormonal symptoms can be compounded by effects resulting from hypothalamic injury, including temperature intolerance/dysregulation, weight gain, and behavioral disturbances.
Diagnosis and Evaluation
Physical Examination
A detailed neurologic examination and history are always important, with one-fourth of patients presenting with hydrocephalus and nearly half presenting with signs referable to increased ICP. Attention should be paid to evidence of endocrinologic or visual dysfunction in the history (see list). Growth arrest is particularly common in children with craniopharyngioma .
Findings Suggestive of an Intracranial Lesion
Headache in the early morning hours or awakening patient from sleep
Vomiting
Headache of less than 6 months duration
Confusion or behavioral changes
Abnormal neurologic examination findings
(Positive correlation between number of predictors and risk of surgical lesion)
On examination, findings may be present secondary to (1) local effects (focal weakness, visual changes, etc.), (2) increased ICP (papilledema, increased head circumference, etc.), or (3) high flow (dilated scalp vessels, bruit on auscultation, cardiac failure).
“Red Flags” on Examination or History
Bradycardia, hypertension, decreased respirations (Cushing response)
Dilated pupil, hemparesis (Uncal herniation)
Fixed downward gaze (Parinaud’s syndrome)
Lethargy, tense open anterior fontanelle in infants
Ataxia with nausea and vomiting
Sudden onset of a third nerve palsy, including involvement of the pupil (appearing dilated)
Sudden onset of severe headache
Imaging Evaluation (13.2)
MRI
MRI is particularly useful for identification of tumors and delineation of the relationship of the tumor to surrounding neurovascular structures including the carotids and their branches, optic apparatus, pituitary gland and stalk, and the hypothalamus/ventricular system (Fig. 13.2). The tumor may appear heterogeneous, with cystic components bright on T1 and T2 and solid portions of the tumor exhibiting variable enhancement following administration of contrast (Fig. 13.2). Obtaining an MRI with axial, sagittal, and coronal planes with and without contrast is critical to preoperative planning and postoperative follow-up. Increasingly, magnetic resonance angiography (MRA) is helpful in delineating vascular anatomy for these same reasons.
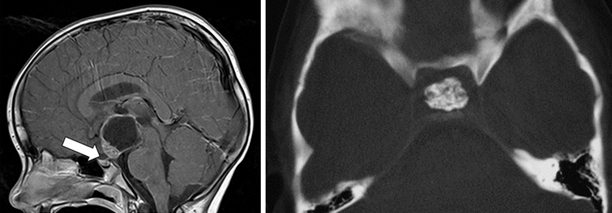
Fig. 13.2
Radiographic appearance of craniopharyngioma. The T1 sagittal enhanced image (left) reveals the large, cystic lesion in the suprasellar region, extending up toward the third ventricle with mixed imaging characteristics. The axial noncontrast CT study (right) demonstrates a solid calcified core in another tumor, a finding of substantial utility in formulating an appropriate treatment strategy
CT
CT is important in the diagnosis and surgical planning for craniopharyngiomas. A distinguishing feature of these tumors is the presence of calcium, which may be difficult to detect on MRI (Fig. 13.2). Regions of calcification are present in the majority of pediatric tumors (up to 90 %) and more than half of adult lesions [10, 17, 22]. Preoperative radiographic visualization of solid calcium deposits (when present) is invaluable to the surgeon in determining the feasibility of specific operative approaches. Moreover, CT is helpful in ascertaining the degree of pneumatization of the sphenoid, ethmoid, and frontal sinuses—relevant to transsphenoidal approaches for sellar tumors (sphenoid), drilling down the planum sphenoidale for frontal approaches (sphenoid/ethmoids), and bifrontal approaches for suprasellar tumors (frontal).
Laboratory Data
Standard preoperative laboratory studies (complete blood count (CBC), clotting times (PT/PTT), type and cross (T&C) for blood bank, chemistry panel (Chem 7)). Replacement of deficient hormones will be performed as needed, with particular attention to cortisol deficiency—patients with craniopharyngioma should be considered in need of supplemental stress dose steroids perioperatively . Replacement of corticosteroids frequently averts disaster in these cases (one can administer dexamethasone 1–4 mg/IV, although hydrocortisone 30 mg/m2 is also effective).
Laboratory Tests for Endocrinology Evaluation
Prolactin
T4, THBR, TSH, Free T4
IGF-1, IGFBP-3
Cortisol (if not receiving steroids)
DHEA-sulfate over age 6 years (if not receiving steroids)
FSH, LH
Estradiol (if female)
Testosterone (if male)
Electrolytes, BUN, creatinine, and serum osmolality
Bone age (at least by the time of hospital discharge)
Other Investigations
When possible, a formal assessment of visual fields and an ophthalmologic examination should be performed prior to surgery [18, 20]. This not only establishes a baseline, but also may help to guide the surgical approach if one optic nerve is substantially impaired and the other has retained function.
Differential Diagnosis
The differential diagnosis of tumors in the hypothalamic/sellar region is large and includes congenital lesions (Rathke’s cleft cyst, arachnoid cyst, dermoid/epidermoid, hypothalamic hamartoma), tumors (pituitary adenomas, germinomas/non-germinomatous germ cell tumors, lymphoma, meningiomas, schwannomas of the cranial nerves, optic gliomas), vascular lesions (aneurysm, cavernous malformation), and inflammatory conditions (neurosarcoid, lymphocytic hypophysitis) [9, 10, 23]. The characteristic calcification and cystic regions of craniopharyngiomas (especially in children) often help substantially with confirming the radiographic diagnosis.
Treatment
Goal
The primary objective of craniopharyngioma treatment is the restoration and preservation of neurologic function with minimal morbidity . There is substantial debate regarding the most effective method for achieving this goal. The difficulty in developing definitive guidelines for care stems in large part from the variability in presentation, the need for exceptionally long follow-up to assess efficacy, and the rarity of these tumors. The preferential use of specific treatments—radical surgery, subtotal resection, radiation, intracystic administration of chemotherapy—may be influenced by both published data and institutional bias. Here we present an overview of surgical techniques used in the management of craniopharyngioma—approaches that will be of use for both radical and subtotal resections.
Stabilization
Access: Large bore IV (at least 2), arterial line, bladder catheter (airway intubation if unable to protect airway), and nasogastric tube if intubated.
Steroid replacement if needed.
ICP control—external ventricular drain if hydrocephalus.
Preparation for Definitive Intervention, Nonemergent
Prior to undertaking an operation, it is critical to define the goals of surgery. An important distinction is whether the objective is a complete resection or a planned subtotal debulking. This topic is controversial and data exist arguing in support of both strategies [1, 6, 18, 24–29]. The policy at our institution is to attempt a total resection at initial presentation in most cases, but exceptions are made when preoperative imaging suggests that surgical morbidity would be unacceptable [18].
Surgical Therapy
Timing
If a child presents with symptoms of increased ICP then urgent operation may be necessary, although it is often possible to temporize with an external ventricular drain and steroids. Given the complex anatomy in the location of these tumors, lengthy operations are common and performing surgery electively is preferable if possible.
A primary surgical goal is complete removal of the tumor, with the best opportunity often present at initial operation. It is important to understand that there can be several equally valid approaches that may be efficacious for a given tumor.
Approach
The growth pattern of an individual tumor can direct a surgeon to a particular approach. In general, craniopharyngiomas fall into three distinct groups: suprasellar/prechiasmatic, suprasellar/postchiasmatic, and sellar. When viewed through this lens, it can be helpful to pair the common approaches (subfrontal, pterional, subtemporal, transcallosal/transventricular, and transsphenoidal) with specific growth patterns to maximize access to the tumor.
Potential Surgical Approaches Based on Craniopharyngioma Growth Patterns
Suprasellar
Primarily intraventricular—Transcallosal/transventricular
Prechiasmatic—Subfrontal
Pterional
Postchiasmatic—Subfrontal
Pterional
Subtemporal
Sellar—Transsphenoidal (microscopic/endoscopic)
Specific Approaches
Subfrontal
The subfrontal approach allows for excellent visualization of optic nerves, carotids, and lamina terminalis. This approach is useful for most craniopharyngiomas, albeit less so for isolated sellar lesions. It affords a wide range of access to the suprasellar region and can easily be combined with other approaches such as pterional, transcallosal/intraventricular, and even sellar (with drilling of the planum sphenoidale). The approach can be unilateral or bilateral, depending on the anatomy of the tumor. For retrochiasmatic and intraventricular tumors, removal of the superior orbital rim and roof improves visualization while minimizing retraction on the frontal lobes (Fig. 13.3).
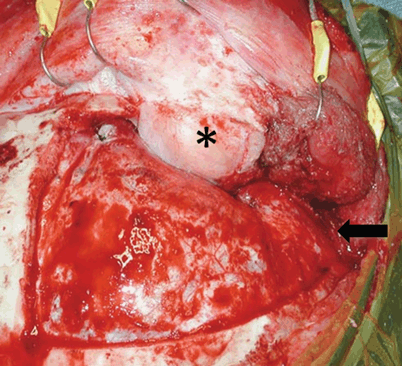
Fig. 13.3




Operative approach (extradural). A unilateral subfrontal approach is demonstrated, with removal of the orbital roof (*). In addition, the craniotomy has been extended inferiorly in order to provide an element of a pterional approach (arrow), highlighting the versatility of these approaches and the importance of preserving flexibility in the operating room
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
