66 Congenital Anomalies of the Kidney and Urologic System
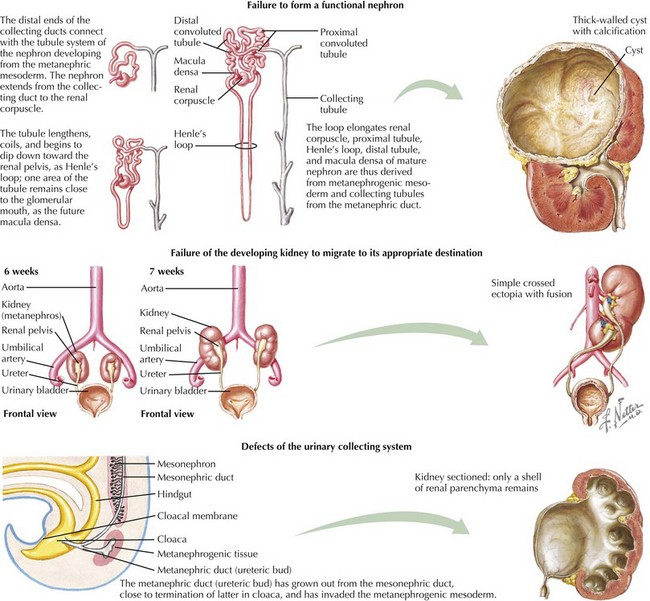
This chapter reviews several common congenital and inherited anomalies of the kidney and urologic system. Congenital abnormalities are a frequent cause of renal failure in children, accounting for more than 30% of end-stage renal disease (ESRD). Congenital anomalies can be subdivided into three categories based on the stage of the primary abnormality in renal embryologic development (Figure 66-1). The first category refers to a failure to form a functional nephron, leading to renal parenchyma malformations, such as renal agenesis or cystic dysplasia. The second group of defects relates to a failure of the developing kidney to migrate to its appropriate destination. This may lead to renal ectopy (pelvic or thoracic kidneys) or fusion abnormalities, such as a horseshoe kidney. The third category describes defects of the urinary collecting system, such as double ureters or posterior urethral valves (PUVs). Here, changes of the renal parenchyma (hydronephrosis, dysplasia) are often secondary to obstructive uropathy or urinary reflux disease. Inherited conditions such as the polycystic kidney diseases (PKD) are the result of specific genetic mutations that may present with detectable renal abnormalities at birth or may not develop until later in life. The following sections detail common and exemplary conditions representative of these subcategories.
Polycystic Kidney Disease
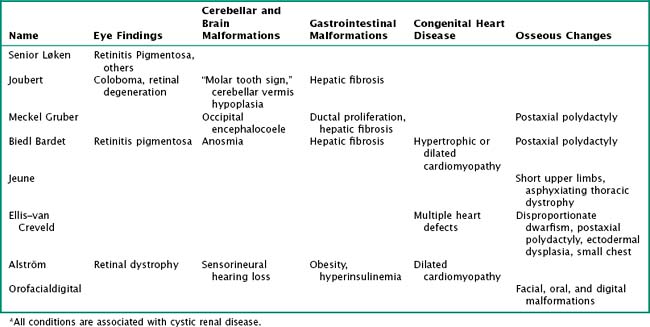
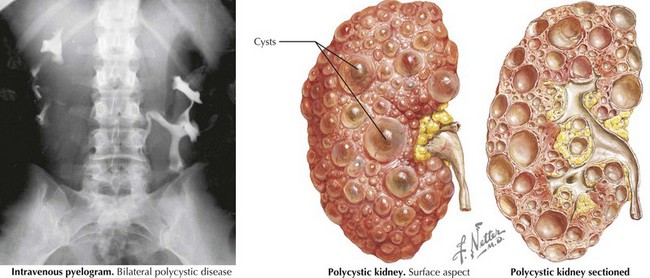
PKDs are a group of genetically inherited conditions in which cyst formation and renal parenchymal replacement can occur at anytime from fetal life to adulthood. There are two major forms, autosomal dominant PKD (ADPKD) and autosomal recessive PKD (ARPKD) (Table 66-1). In addition, a number of rare pleiotropic disorders exist, which are loosely associated because of similar clinical and pathophysiologic features, including renal cystic and hepatobiliary disease (Table 66-2). All of these disorders share ciliary dysfunction as a common principle in their pathogenesis. PKD proteins have been localized to the cilia or basal body, and loss or abnormalities of cilia in the kidney are associated with cyst development (Figure 66-2).
Table 66-1 Autosomal Recessive Polycystic Kidney Disease versus Autosomal Dominant Polycystic Kidney Disease
| Autosomal Recessive Polycystic Kidney Disease | Autosomal Dominant Polycystic Kidney Disease | |
|---|---|---|
| Gene | Chromosome 6p21.2-p12 | Chromosome 16p13.3, chromosome 4q13-q23 |
| Protein | Fibrocystin | Polycystin 1, polycystin 2 |
| Age of presentation | Commonly prenatally, childhood, adolescence | Highly variable |
| Renal cysts | Radial pattern | Anywhere in kidney, varying size |
| Extrarenal manifestations | Biliary obstruction, hepatic fibrosis with portal hypertension, liver failure | Liver cysts, pancreas cysts, vascular cysts (“Berry aneurysms”) |
| Things in common | Renal cysts do not communicate. Disease may present at any age, including prenatally. Gene defect leads to malfunction of the ciliary body. The kidneys are usually enlarged. Usually bilateral disease (exception: early diagnosis ADPKD in childhood with positive family history) | |
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


