Types of enzyme blocks and their consequences in CAH. The inherited defect in steroid biosynthesis of cortisol decreases cortisol, which leads to an increase in ACTH. This can result in compensated cortisol block and precursors (androgens, corticoids).

Deficiency of 21-hydroxylase enzyme defect. The potential spectrum of salt loss and shock, virilization, amenorrhea, and infertility.
The incidence of classic 21-hydroxylase deficiency CAH ranges from 1 in 5000 to 1 in 16000 live births [1,7]. Approximately 1 in 60 people is a carrier for classic CAH [1]. Interestingly, CAH is more common in white and Hispanic people than in African Americans in the USA [1,7]. The incidence of nonclassic 21-hydroxylase deficiency CAH, however, is higher than that of classic 21-hydroxylase deficiency, and the incidence is even as high as 3.7% in Ashkenazi Jews and 1.9% in Hispanics [8].
Patients with CAH will need lifelong treatment and care. For newborns and infants, the treatment goals are to prevent an adrenal crisis, to assign a gender with surgery in the more severe cases of CAH, and to protect linear growth [5]. For children and adolescents, the treatment goals are to protect linear growth, to promote a normal pubertal development, and to maintain an appropriate body weight. Lastly, the treatment goals of adults include fertility, prevention of metabolic syndrome, and prevention of osteoporosis.
Pathophysiology
Although CAH is an autosomal recessive disorder, its pathophysiology is more complex than most. There are three main steroids produced by the adrenal: glucocorticoids, mineralocorticoids, and sex steroids. Production is controlled by adrenocorticotropic hormone (ACTH). ACTH is regulated by the central nervous system, and its production is increased during times of physiological stress with corticotropin-releasing factor (CRF) from the hypothalamus. ACTH and CRF are then controlled by negative feedback from circulating plasma levels of cortisol. ACTH augments StAR protein function, which moves free cholesterol to the inner mitochondrial membrane so that pregnenolone is produced and steroidogenesis can occur.
Thus, when cortisol is not present because of CAH, ACTH continues to be produced and steroidogenesis continues unchecked in the metabolic pathways not affected by CAH and with an accumulation of precursor molecules in the metabolic pathways affected by CAH. The precursor molecules (such as 17-hydroxyprogesterone, progesterone, and androstenedione) that have accumulated due to an enzyme deficiency from CAH are then used in the metabolic pathways not affected by CAH and more androgens are created. Additionally, the excess ACTH causes hypertrophy in the adrenal glands of the zona fasciculata and zona reticularis, and thus, the name CAH.
Clinical presentation
The internal genitalia of a woman with CAH remain normal because she does not have müllerian-inhibiting hormone from testicular Sertoli cells [4]. Her external genitalia, however, can appear normal at birth or ambiguous/virilized, depending on the type and severity of her CAH (Figure 11.3a–c) [9]. Additionally, excess ACTH can cause increased pigmentation of nipples and genitalia [5]. If the patient’s CAH is not appropriately treated, she will continue to have excess exposure to androgens. This continued excess exposure can lead to progressing penile/clitoral enlargement, premature development of pubic and axillary hair, acne, and apocrine odor (Figure 11.3a–c) [1,4]. While an individual with CAH may appear tall as a child, she will often be short as an adult due to premature epiphyseal fusion from accelerated skeletal maturation [1].
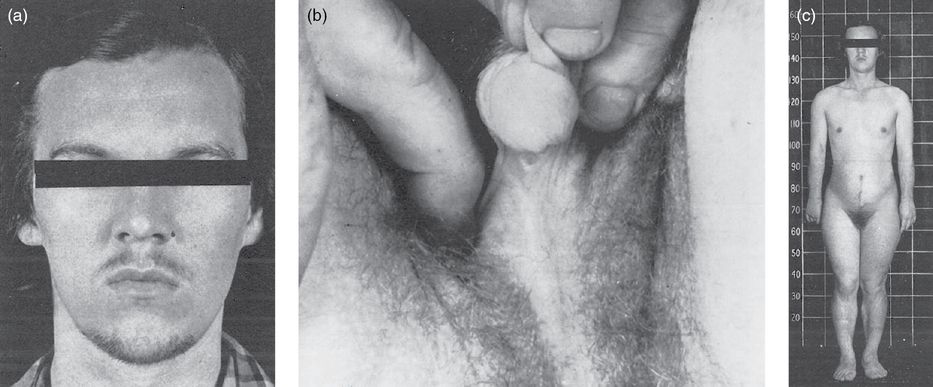
(a) Patient N.W. at age 16 after 3 days without shaving. (b) Patient N.W.’s external genitalia before surgery. (c) Patient N.W. after 8 months of cortisone therapy; notice the breast development. Reproduced with permission from Jones HW Jr., Scott WW. Female Intersexuality with Adrenal Hyperplasia. In: Jones HW Jr., Scott WW (Eds.) Hermaphroditism, Genital Anomalies and Related Endocrine Disorders. The Williams and Wilkins Company; 1971: Ch. 9.
If a CAH patient is successfully treated from birth, she will likely have a relatively normal pubertal age with normal appearance of secondary sex characteristics [4]. If she is not successfully treated, hyperandrogenism can result. Hyperandrogenism in adult females can appear as alopecia, acne, infertility, hirsutism (Figure 11.3a–c), and absent/irregular menses [1,4]. Infertility in these female patients can be due to various etiologies, including anovulation, irregular menses, abnormal introitus, and uncontrolled progesterone levels. There is also some research demonstrating an association of CAH with polycystic ovary syndrome (PCOS) [10].
21-Hydroxylase deficiency
Female patients with the classic form of 21-hydroxylase deficiency will suffer from the more severe hyperandrogenism symptoms in utero, virilization of external female genitalia, as a result of androgen exposure from about the sixth week of gestation (Figure 11.4) [1,4]. This virilization can result in genital ambiguity in females upon birth, ranging from clitoromegaly to a penile urethra. There are two subtypes of classic 21-hydroxylase deficiency CAH: a simple virilizing form and a salt-wasting form, the latter related to the adequacy of aldosterone production (Figure 11.2). Over 75% of classic 21-hydroxylase deficiency CAH presents with the salt-wasting subtype, which is the more dangerous as it subjects the newborn to risk of adrenal crises [4,7]. For patients with salt-wasting classic 21-hydroxylase deficiency, less than 1% of enzyme activity remains [5].
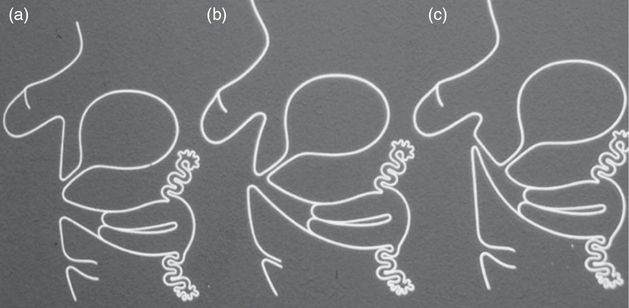
External genitalia as dependent on exposure to androgens. (a) Female external genitalia exposed to testosterone after 12th week of pregnancy, leading to clitoral hypertrophy. (b) Earlier exposure between 7 and 12 weeks, leading to progressive masculinization of urogenital sinus. (c) Exposure at 7 weeks leading to penile urethra.
Patients with the nonclassic form of 21-hydroxylase deficiency CAH are only mildly affected by their enzymatic deficiency and tend to present as adults with signs of hyperandrogenism, if they present with signs at all [4]. They can present with premature development of pubic hair, advanced bone age, severe cystic acne, infertility, alopecia, hirsutism, voice deepening, and male habitus. Their menarche tends to be normal or delayed, although secondary amenorrhea frequently occurs. Both classic and nonclassic 21-hydroxylase deficiency patients can exhibit poor growth if attempts at treating them with glucocorticoids exceed the appropriate physiological levels [4].
Diagnosis
Early diagnosis of CAH is important to improve morbidity and mortality. Female infants with classic 21-hydroxylase deficiency are often diagnosed by ambiguous external genitalia at birth and/or salt-wasting (i.e., failure to thrive, hypovolemia, hypotension, hyponatremia, and hyperkalemia) [5]. After identifying the ambiguous genitalia, pelvic ultrasound and genitograms are appropriate to evaluate the newborn [11]. Children with virilizing classic 21-hydroxylase can present with precocious adrenarche and accelerated growth. Women with nonclassic 21-hydroxylase deficiency tend to present the same as women with PCOS. For that reason, in patients who present with hyperandrogenism and are diagnosed with PCOS, it is necessary to consider CAH as well [4].
Hormonal diagnosis
CAH can be diagnosed by evaluating a female’s hormone levels with radioimmunoassays, enzyme-linked immunosorbent assays, and time-resolved fluoroimmunoassays. For classic 21-hydroxylase deficiency, a high concentration of 17-hydroxyprogesterone (17-OHP), which is the precursor of the enzyme that is defective, is diagnostic [1,4]. Blood should be drawn in the early morning for this assay [12].
For nonclassic 21-hydroxylase deficiency, a corticotropin stimulation test (CST), also known as the acute ACTH stimulation test, is diagnostic. For a CST, 250 μg of cosyntropin (Cortrosyn) is given intravenously, then 17-OHP and Δ4-androstenedione are measured at baseline and then again at 30–60 minutes. Because it may be impractical to do a CST on all women with hyperandrogenism signs, it has been suggested that unstimulated 17-OHP levels (170–300 ng/dL), measured in the morning and in the follicular phase of the menstrual cycle, be used to screen women [1]. Additionally, plasma renin activity should be measured in order to determine whether the patient also has a mineralocorticoid deficiency.
Molecular genetics
Although genetic testing may be useful, it is not the gold standard for diagnosis of 21-hydroxylase deficiency and should only be used when 17-OHP and CST results are equivocal [12]. There are over 120 mutations on CYP21A2 associated with CAH, which result in partial to total loss of enzyme activity [1]. The various forms of CAH tend to have specific genotypes; however, phenotypes do not always correlate with the genotype [1,4]. For CAH, a recessive disease, usually it is the least deficient mutation of the two alleles that governs. Thus, for classic 21-hydroxylase deficiency, usually two alleles with severe mutations occur together. For nonclassic 21-hydroxylase deficiency, either the patient has two mild mutations or one severe and one mild mutation on her alleles. The screening panels that are commercially available tend to only measure the 10–12 most common mutations [1]. Genetic analysis can be limited because of the complexity of CAH gene duplications, gene deletions, and gene rearrangements within chromosome 6p21.3 [12].
Newborn screening
All 50 states in the USA screen for CAH [12]. Screening for CAH involves measuring 17-OHP, and often detects newborns with classic 21-hydroxylase deficiency. Of note, female newborns with classic 21-hydroxylase deficiency will often have genital ambiguity due to virilization as another indicator of CAH. The Endocrine Society recommends that all newborns are screened for 21-hydroxylase deficiency with an initial immunoassay and then further evaluation of positive immunoassays with liquid chromatography/tandem mass spectrometry [12]. Nonclassic 21-hydroxylase deficiency is often not detected by newborn screening because their 17-OHP levels tend to be lower [1]. There has been some research advancing the use of ultrasonography to screen newborns for enlarged adrenal glands in order to achieve more rapid diagnostic results than with biochemical assays [13].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


