•S upervise : all aspects of the trial are ultimately the responsible of the investigator
•M aintain Records : meticulous record keeping is critically important
•A dhere to the protocol : deviations from the protocol should not be implemented without the notification and consent of the sponsor
•L earn the investigator brochure : provides a summary of all known information about the compound under study
•L et the FDA inspect : to determine whether the study has been conducted according to GCP
•Re P ort adverse events : the investigator is obligated to report all AEs regardless of the perceived relation to the study drug
•R etain records : case report forms and all regulatory documents
•I nform subjects : of all aspects of the trial before enrollment in comprehensive language and notify of all changes
•N otify the IRB
•T rain staff
National Institutes of Health (NIH): The NIH, founded in 1887, serves as the focal point for federal funding of medical research in the United States. The NIH has the goal of acquiring the knowledge required to prevent, detect, diagnose and treat disease and disability. The NIH accomplishes this goal through research in its laboratories, support of research in academic and research institutions and hospitals, through grants, and through the training of scientists, researchers and physicians through grants. The NIH remains the greatest contributor to medical research and a major player in drug development. The NIH is also the sponsor and administrator of the clinical trials.gov website, where ongoing trials are posted.
Institutional Review Board (IRB) and Ethics Committee: The IRB within the US and the Ethics Committee outside the US are charged with insuring that research is conducted in a safe, scientifically sound, and ethical manner. The IRBs are established and governed by regulations outlined in the United States Code of Federal Regulations [12], which is administered by the Office of Human Research Protections. These regulations are based on the ethical principles set forth in the Belmont Report [15] that include respect for the research participant, beneficence for society at large, and justice with equal access to participation and equal treatment of subjects in a research study. Also outlined in these regulations are requirements for the informed consent that are listed in Table 43.2.
Sponsor: The sponsor is a company or organization providing resources that enable the trial to proceed. A sponsor’s contribution will range from providing the agent to be studied, to providing full financial and logistic support for the development program.
CROs: A CRO is an organization hired to assist with implementation of the clinical trial by the sponsor. The CRO’s roles and responsibilities vary but often include responsibility for negotiation of the contracts with the sites, for negotiating the exact language of the informed consent form (ICF), for monitoring of sites, and for data collection. In some cases, the CRO will hold and lock the database and are responsible for data analysis.
Data Monitoring Committee (DMC): A DMC may serve many roles; the DMC can function solely as a Data and Safety Monitoring Board or be involved in endpoint analysis and in trial modification. Key questions you should ask yourself are: “Would a DMC help mitigate risk to subjects? Would a DMC help maintain study integrity?” If yes, a DMC is indicated.
Drug Development Process
Reviews over the past decade have emphasized the large number of chemical compounds that must be examined as potential drugs during the development of each successfully approved agent. It has been estimated, that up to 10,000 compounds are examined for each therapeutic agent approved. The drug development process is also time consuming with drug development taking up to 16 years from the identification of a likely target to the approval of a drug effective against the target [16].
The development of every new drug begins with an idea. Often this idea or insight comes from a clinical observation, such as the recognition of a drug’s “side effect” during drug development for another indication, such as the observation of hair growth in subjects receiving a drug for hypertension. The idea or insight also often comes from advances in our understanding of disease pathophysiology. For example, the basic research that established the critical role of cytokines in inflammation, and the clinical research that confirmed that cytokines were elevated in inflammatory bowel disease (IBD), led to selection of tumor necrosis factor alpha as a target and to the development of anti-TNF agents for treatment of IBD. More recently the recognition of the role of IL-12 and IL-23 and IL-17 in inflammation has led to ongoing exploration of multiple targets in inflammatory diseases. The idea or insight leads to selection of a target, and the development of an agent effective against the target.
Drug development is generally divided into four stages: drug discovery, preclinical studies, clinical studies and regulatory review. As outlined in Fig. 43.1 it is estimated that nearly half of the time invested in drug development is spent in the drug discovery and preclinical stages (6.5 years), with the remainder spent in clinical trials (7 years) and in review by the regulatory agencies (1.5 years).
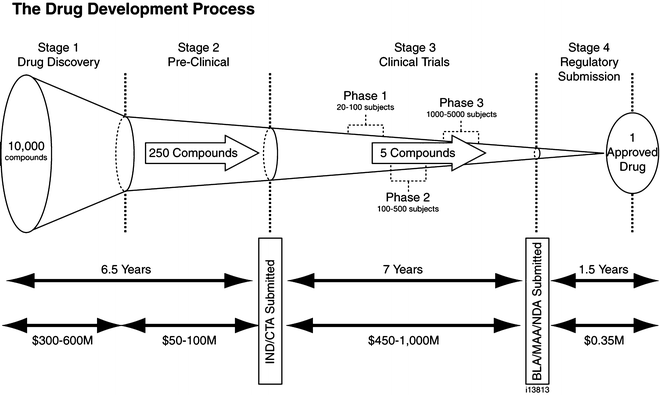
Fig. 43.1
The drug development process
During the first stage, drug discovery, compounds are evaluated for their effectiveness against identified targets using in vitro assay systems and, when available, animal models. Once a promising candidate has been identified, that candidate is further examined in the preclinical stage with toxicology studies to evaluate the potential of the drug for mutagenicity/genotoxicity, teratogenicity and carcinogenicity. Pharmacological studies in animals are also undertaken in the preclinical stage to establish the pharmacokinetic (PK) parameters that define the absorption, distribution, metabolism, and excretion of the agent.
This preclinical toxicology data is combined with a clinical development plan to form an independent new drug (IND) application in the United States, or a clinical trial application (CTA) in European countries,
The IND is submitted to the FDA and/or CTA are submitted to the local health authority (the FDA in the US) at the end of preclinical studies and prior to the first in human phase 1 trial. The IND and CTA applications provide the reviewer at the health authority with the data required to ensure the potential safety of the candidate compound and provides the basis for clinical studies. The health authority (FDA, in the US) must approve the CTA (or IND) prior to trials in man.
The clinical study stage is divided into four phases.
In phase 1 trials; healthy volunteers are given the agent to confirm the PK parameters in man, while evaluating the safety of first single doses and then multiple doses of the agent. Generally restricted to a limited number of subjects (20–80), phase 1 trials often require hospitalization to enable the blood sampling required for the PK analysis.
In phase 2 trials; initial efficacy and safety data are collected in subjects with the disease under study. Generally multiple doses will be used during these studies and larger numbers of subjects are enrolled (250–400). Once the phase 2 studies have established the safety and efficacy of the drug, a full development decision is made, a post phase 2 meeting is scheduled with the FDA, and trials are designed for Phase 3 to establish the efficacy and safety of the drug using dose regimens planned for the clinical use.
In phase 3 trials; well-defined selection criteria are used (inclusion and exclusion criterion) and multiple trials are conducted to establish the efficacy and safety of the drug under study. Phase 3 trials require a larger number of subjects (1,000–5,000) utilizing dose regimens proven to be effective in the phase 2 trials. The FDA generally requires independent confirmation of efficacy with two adequately powered and controlled trials.
In phase 4 trials; the drug has been approved and the studies are undertaken to further define the drug’s efficacy, safety, and pharmacology.
The new drug application (NDA) submitted to the FDA and the CTA submitted to countries outside the US describe all data collected on a product and includes the chemistry, manufacturing process and quality control, animal toxicology, pharmacokinetics in animals and in man, and the clinical trial evidence on efficacy and safety. The results of phase 1–3 studies form the basis of a drug application (NDA). Only after review and approval of the NDA by the regulatory authority and receipt of a signed approval action letter can a drug be marketed.
Key Clinical Trial Design Features
Though there are many important clinical design features that must be considered during development of a clinical protocol we will review the four key design features of patient population, study design, the choice of endpoint and the dosing regimen.
The patient population to be studied in each trial must be carefully chosen to adequately define the study population. Competing priorities in this process include the need to minimize variability by tightly defining the population under study, while studying a population, which will allow physicians to generalize the results of the trial to the patients in their practice.
The study design chosen must match the hypothesis and the objective of the study. As an example, a classic design when the objective is to establish that an agent is effective in inducing response and remission would compare an active agent to a placebo in a population with active disease. In contrast when the objective is to establish that the agent maintains remission, a study, which randomizes subjects in remission to placebo or active agent and then follows the subjects long enough to ensure exacerbation of disease to occur in the subjects would be a good choice.
The choice of endpoint and the timing of the endpoint measure are key aspects of the trial. The endpoint should be validated and if possible have a history as being accepted by both regulatory authorities and opinion leaders. The choice of the timing of the assessment of the endpoint measure is also critical. In a trial designed to examine response to a drug, if the endpoint is established too early, the drug will not have time to induce response, if too late, placebo response may mask a true effect of the active agent.
The dose and dosing regimen must be chosen to ensure adequate serum levels of the active agent. This is generally based on preclinical PK and is confirmed by phase 1 PK.
The Regulatory Agencies and the Regulatory Process
Since 1938, when the Food, Drug and Cosmetic Act was passed, all new drugs brought to market in the US have required the approval of the FDA through the NDA process [17]. Though initially this review was restricted to ensuring the safety of new drugs, the NDA process was modified in 1962 with the Kefauver-Harris Amendments to the 1938 Act to add efficacy for intended use and confirmation that the benefits of the drug outweigh its know risks to the review of the NDA [12,15,17]. The FDA Modernization Act reduced review times, allowed for electronic submissions and proposed the first “pediatric rule” which mandated assessments of new drugs and biologic products for pediatric patients.
Though this first “pediatric rule” was overturned in a federal district court, in Dec. 2003, the Pediatric Research Equity Act (PREA) required pediatric studies for all NDAs and biologics license applications (BLAs) and here by re-established the pediatric rule. In 2002, the Best Pharmaceuticals for Children Act established a voluntary program that awards a 6-month exclusivity incentive to sponsors of marketed products that conducted pediatric studies in response to a written request from the FDA. The BPCA was the result of a partnership between the FDA and the NIH to promote pediatric studies for off-patent products.
In the EU, the Pediatric Regulation in Europe in effect since January, 2007 established a framework of rewards, incentives and obligations for pharmaceutical companies. The goal of this regulation was to encourage the development of medications for children which had been appropriately tested, authorized and formulated for use in children. The pediatric regulation established both a pediatric committee (PDCO) at the EMA, and a requirement for a pediatric investigation plan (PIP). The PIP is a binding roadmap that details the timing and measures proposed to demonstrate quality, safety and efficacy in the pediatric population. It specifies which population subsets need to be studied, by what means and by what date. The regulation specifies that the PIP should be submitted no later than upon completion of human PK studies in adults, potentially prior to Phase 2 Proof of concept trials. An approved PIP is required prior to a marketing authorization application filing for adult indications.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


