Philip T. Levy, Amish Jain Key points Chronic pulmonary hypertension (cPH) in neonates is a heterogeneous disease process characterized by an elevation of pulmonary artery pressures (PAP) and prolonged exposure of the right ventricle (RV) to high afterload that collectively contribute to higher morbidity and mortality.1–4 The diagnosis and management of the hemodynamic status of preterm infants with cPH can be challenging, owing to the multitude of etiologies and the unique characteristics of the pulmonary circulatory system. The causes of neonatal cPH may be described based on endotype of the pulmonary vasculature.1,2,5 These include conditions associated with maladaptive pulmonary vasculature (e.g., chronic lung disease [CLD], pulmonary hypoplasia and genetic conditions like trisomy 21), those associated with maldeveloped pulmonary vasculature, and those associated with congested pulmonary vasculature caused by high pulmonary blood flow (PBF), or by raised pulmonary capillary wedge pressure (PCWP). However, physiologically, the spectrum of neonatal cPH can be understood from the direct relationship between mean PAP (mPAP) and pulmonary vascular resistance (PVR), PBF, and PWCP, defined by the following equation: mPAP = (PVR × PBF) + PCWP.1 Traditionally, cPH has been thought to originate from a secondary progressive rise in PVR beyond the first month of age (often with initial successful postnatal transition) and is seen most frequently as a secondary complication with CLD. High mPAP from exposure to high PBF, most commonly in the context of atrial-level left to right shunt, or a combination of high PBF and PVR, is not uncommon. Less frequent and currently not fully understood, high PCWP conditions may also contribute to cPH in this patient population. Chronically elevated mPAP increases afterload on the immature right ventricle (RV), which may lead to ventricular remodeling, RV congestion (dilatation), and in severe cases RV systolic dysfunction and poor cardiac output. A comprehensive clinical and echocardiography assessment focused on evaluating the major determinants of mPAP, possibly combined with echocardiography measures of myocardial performance and cardiac biomarkers, and with judicious use of invasive diagnostic modalities, can help define underlying etiologies. This approach will provide hemodynamic profiles of the disease in patients, allowing clinicians to adopt a physiology-driven individualized management plan.1,2 This is important as recent pilot data also indicates an association between neonatal cPH and impaired growth and neurodevelopment during early childhood in preterm infants, after accounting for CLD severity.6 This chapter discusses the risk factors, underlying etiopathologies, and hemodynamic assessment of cPH in neonates and provides a physiology-based approach for the diagnosis of the variability of the presenting phenotypes of neonatal cPH with special considerations based on new recommendations and emerging diagnostic methods. Although term-born infants can also develop cPH in association with chronic lung disease, congenital heart disease (CHD), and structural and genetic abnormalities of the airways, pulmonary vasculature, or parenchyma,1 the focus of this chapter is cPH in preterm neonates. That population accounts for the greatest proportion of patients seen in pediatric pulmonary hypertension clinics. Pulmonary hypertension can present in premature infants at different stages of initial hospital stay with varying hemodynamic signatures, including acute and early (e.g., delayed perinatal transition with persistent elevation of PVR), acute and late (associated with acute complications such as sepsis and pulmonary hemorrhage), and chronic and late (i.e., cPH), which is typically sustained well beyond the initial hospital stay.7 The chronic and late form of pulmonary hypertension commonly lasts into infancy and early childhood and may have pulmonary vascular ramifications across the lifespan. Extremely premature neonates, born <28 weeks’ gestational age, are particularly vulnerable to developing cPH and have been the focus of the majority of research to date. In preterm infants cPH is intricately linked, though not always, to the occurrence and severity of CLD.8–15 While the overall incidence of cPH in this patient population is ∼20%, the reported range is 14–44%, depending on the severity of underlying CLD.4,16–18 Interestingly, even infants with mild or no CLD have some risk for pulmonary vascular disease and overt cPH.8–12,19 Recent evidence indicates that 2–20% of infants born less than 30 weeks’ gestation without BPD may develop evidence of pulmonary hypertension during the neonatal period.3 However, clinical observations suggest that this disease, at least in the short term, may not be as aggressive and pathological as overt cPH associated with moderate to severe CLD. Several prospective and retrospective cohort studies have now confirmed that, compared to CLD without cPH, infants with cPH are at much higher risk of in-hospital and post-discharge mortality as well as respiratory morbidities, such as duration off mechanical ventilation and home oxygen therapy.3,9,11,13,15,20–22 Among preterm neonates with cPH diagnosed at 36 weeks postmenstrual age (PMA), the rate of reported associated mortality ranged from 20 to 50% and was linked closely with the severity of cPH.18,23 Among infants with evidence of supra-systemic pulmonary pressures at 36 weeks PMA, the mortality rate may be as high as 60–70%, with most occurring within the first 6–8 months after diagnosis.18,23 Among survivors though, the natural history of cPH appears to follow the pattern of CLD itself, where the disease commonly resolves with growth over 36 to 42 months.23 However, physiologically, echocardiographic evidence of subclinical relative pulmonary vascular disease persists in neonates with CLD well into late childhood and beyond.24–26 Further, the burden of cPH may extend beyond the neonatal period and is associated with respiratory diseases during early childhood,21 poor growth,27 and sub-optimal neurodevelopment.3,21 Maternal and fetal risk factors can affect the development of pulmonary vasculature and how it adapts to extra-uterine life. Physiologically, reduced vascular branching, altered pattern of vascular distribution, disruption of vascular signaling pathways, endothelial injury, and abnormal smooth muscle proliferation are some of the underlying mechanisms of the progressive increase in PVR.28 Exacerbating factors include air trapping induced abnormal stretch of small pulmonary arteries, atelectasis leading to constraining pulmonary vessels, and acute episodes of hypoxia and hypercarbia, all of which can induce pulmonary artery vasoconstriction and altered vessel morphology in premature neonates.15 These can lead to injury of the lung parenchyma and affect the molecular pathways responsible for increased pulmonary vasomotor tone, leading to cPH.29,30 Epidemiologically, the known risk factors contributing to development of cPH in preterm neonates may be seen as demographic variables, which are unlikely to be amenable to interventions, and postnatal acquired variables, which clinicians may have an opportunity to alter (Figure 26.1).31,32 The degree of prematurity remains the most consequential risk factor for developing cPH in preterm neonates.33 Other demographic factors include poor intrauterine growth, pulmonary hypoplasia–related conditions such as prolonged oligohydramnios, and factors promoting in utero pulmonary vascular remodeling, such as maternal hypertension and diabetes, antenatal closure of ductus arteriosus, twin-to-twin transfusion syndrome, chylothorax, and hydrops fetalis. Acquired postnatal factors may include known risk factors for severity of CLD, such as hyperoxia, duration of mechanical ventilation, inflammatory conditions like sepsis, necrotizing enterocolitis and ventilation-associated pneumonia, chronic aspiration, chronic exposure to shunts resulting in high pulmonary blood flow (large patent ductus arteriosus, atrial, or ventricular septal defects), and cumulative burden of hypoxemia by virtue of activation of hypoxic pulmonary vasoconstriction. Further, occurrence of pulmonary edema is both a contributory factor to worsening of cPH and a consequence of cPH itself, as fluid leaks into the lung interstitial space due to high intravascular pressure. The subsequent rise in interstitial pressure causes further obliteration of pulmonary microvasculature and redistribution of blood to even smaller vascular compartments, raising intravascular pressure. In addition, early echocardiographic evidence of pulmonary vascular disease has been identified as a risk factor for subsequent development of cPH by 36 weeks PMA.8,9,11,21,34 Levy et al.,9 utilizing deformation imaging, demonstrated that changes in ventricular septal strain patterns in the first postnatal week were associated with a higher risk of cPH (RR 2.15; 95% CI 1.18–4.33). Mourani et al.,8,21 in a large prospective cohort, observed that ventricular septal wall flattening at 7 days of age was associated with increased risk of the subsequent diagnosis of cPH at 36 weeks PMA (RR 2.85; 95% CI 1.28–6.33). However, the sensitivity and specificity of early changes was only 62% and 61%, respectively, to identify subsequent cPH cases. In several infants with early echocardiographic signs the pulmonary vascular changes resolved without cPH diagnosis at 36 weeks PMA and in some infants without any early vascular changes, cPH resulted. The optimal timing and diagnostic criteria to identify preterm neonates with significant pulmonary vascular disease, early in the disease course, are still being investigated.35 Preterm infants with BPD are at risk for dysregulation of pulmonary vasculature, characterized by vascular growth arrest. This delicate pulmonary vascular system is further impacted by states of hyperoxia, alveolar hypoxia, and hemodynamic perturbations, which all play a role in proliferation of the smooth muscle and integration of myofibroblasts and fibroblasts into the vessel walls. These vascular changes ultimately lead to elevation of the pulmonary vascular resistance with decreased compliance and vascular narrowing, and ultimately cPH, RV strain, and RV failure. In addition, systemic arterial stiffness will lead to increased afterload, LV hypertrophy and dysfunction, and possibly pulmonary edema. cPH is a disease of altered RV-pulmonary vasculature interactions where the main determinant of the clinical symptoms and prognosis is the progressive response of the RV to pressure loading (Figure 26.2). The challenge for the neonatal RV in cPH is to remain hemodynamically coupled to the pulmonary circulation.36 Initially, the RV adapts to the increasing vascular load by enhancing contractility to maintain PBF, by virtue of the myocardium’s inherent contractile reserve. Ongoing exposure to altered loading conditions then leads to RV remodeling and adaptive responses. An important adaptive mechanism that enhances the contractile capabilities of the RV is muscle hypertrophy with increased wall thickness, referred to as homeotropic adaptation. This is a relatively beneficial adaptation, as it allows the RV to maintain output while keeping its wall stress and oxygen demand low (wall stress is directly related to chamber diameter and inversely to the wall thickness). Sustained and progressive pressure-volume loading of the RV, however, may lead to maladaptive ventricular remodeling in which the RV dilates, referred to as heterotropic adaptation. While a dilated RV can maintain normal cardiac output by virtue of higher end-diastolic volume despite lower ejection fraction, through the Frank-Starling principle, it comes at the expense of significant rise in wall stress and oxygen consumption. At this stage, the end-diastolic pressures in the RV increase, which can then transmit upstream, raising the central venous pressure, clinically resulting in systemic and pulmonary edema (RV congestion or congestive heart failure). The dilated RV also pushes the interventricular septum leftwards, distorting the normal left ventricular geometry,37 which reduces the ability of the left ventricular contraction to support RV output by trans-septal pressure conduction (left ventricular contraction can support up to 40% of RV output), placing further contractile load on the failing RV. As the sarcomeres reach the maximal length corresponding to the plateau of the Frank-Starling curve, further dilation only causes rise in wall stress without any corresponding increase in the cardiac output. This may precipitate progression to end-stage overt RV failure, characterized by severely reduced contractility, uncoupling of RV from the high afterload, critically reduced RV output, and clinically, profound oxygenation failure and shock. In clinical practice most patients demonstrate a combination of homeotropic and heterotropic adaptive response, the relative extent of which may vary between individuals, producing patient-to-patient variability in the clinical phenotype despite similar afterload exposure. Among preterm neonates with cPH, heterotropic adaptation with RV dilatation appears to be the predominant response as the preterm myocardium is often incapable of rapid increase in wall thickness, making them highly vulnerable to the adverse outcome of progressive RV dilatation and failure.38 Furthermore, since the preterm myocardium is composed of underdeveloped contractile mechanisms with disorganized myofibrils, an immature calcium handling system, and inadequately compliant collagen, it has inherently reduced diastolic properties and compliance, making it highly sensitive to pressure/volume loading conditions.39 Further, preterm neonates have a relatively low total pulmonary vascular capacity (less total number of alveoli-capillary units), which is further reduced in the presence of parenchymal disease like CLD, making preterm pulmonary vasculature less tolerant to volume overload and susceptible to getting congested and developing cPH, in the setting of relatively smaller left-to-right shunts than term-born neonates might tolerate. The phenotypic presentation of cPH is governed by slow, sustained exposure to elevated PAP and RV adaptation to it, which is in stark contrast to the typical severe acute hypoxic respiratory failure and hemodynamic lability associated with an acute pulmonary hypertensive crisis, until late stage of RV failure ensues.4,36 Preterm neonates with cPH typically present with more subtle respiratory symptoms such as dependence on respiratory support and oxygen therapy rather than overt oxygenation and respiratory failure. Given the major overlap in clinical symptoms and respiratory course, cPH may be difficult to distinguish clinically from the underlying lung disease (CLD) itself, particularly early in the disease course.4,36 Therefore clinicians are required to be proactive and have a high index of suspicion. Any neonate with chronic parenchymal lung disease where the respiratory course deviates from the expected, such as unexpected and unexplained worsening of oxygenation status late in the hospital course or lack of expected improvement, should be considered for evaluation of cPH. Ongoing pressure-volume overload often leads to RV dilatation and raised central venous pressure, manifesting through a varying combination of signs of pulmonary heart disease i.e., cardiac symptoms and signs occurring secondary to a primary pulmonary pathology. In the context of cPH it indicates occurrence of upstream congestive right heart failure, producing signs such as excessive weight gain, systemic dependent edema, hepatomegaly, pulmonary edema, and/or inability to establish oral feeding due to dyspnea.36 In the late stages overt RV failure will present as progression to profound and refractory hypoxic respiratory failure and systemic shock from severe low RV output. Another possible manifestation and complication of neonatal cPH is an out-of-proportion hypoxemia and acute pulmonary hypertension precipitated by intercurrent illnesses such as sepsis, viral infections, or even urinary tract infections (acute on chronic pulmonary hypertension). It is thus important for clinicians to counsel parents of neonates with cPH before discharge from the hospital about risk of PH crises due to viral illnesses and the need for early evaluation by the pediatric care provider. It is important to communicate the disease course with primary care providers (especially the increased risk of an acute deterioration after a viral illness) and ensure robust follow-up. As described above, optimal diagnosis and monitoring of cPH in preterm neonates requires heightened clinician’s awareness of the problem and a high index of suspicion, liberal usage of noninvasive imaging, along with judicious use of invasive and advanced assessments (Table 26.1).38,40 The gold-standard investigation for confirming diagnosis, pathophysiology, and vasoreactivity of cPH is cardiac catheterization; however, its invasive nature and associated risk for complications makes it an undesirable option for the majority of preterm neonates.41 Given these pragmatic considerations, echocardiography is the most commonly employed modality and is considered the standard of care to screen, establish diagnosis and disease severity, and provide longitudinal follow-up to monitor disease progression and response to interventions,4,42,43 as recommended in current practice guidelines44 and expert opinions.4,36,45–47 Using a comprehensive approach, bedside echocardiography provides relevant hemodynamic information, which, when integrated with the phenotypic clinical presentation, offers a pathway for which to develop a scientifically-based diagnostic impression, determine a pathophysiologically appropriate management strategy, and evaluate response to therapeutic interventions.38 Using a systematic approach, echocardiography-derived measurements may be considered under three broad categories (Table 26.2): (1) qualitative, semi-quantitative, and quantitative assessment of pulmonary pressures (RV afterload assessment); (2) evaluation of right and left ventricular size and pump performance; and (3) appraisal of shunts.2 1Cardiac catheterization is used for the assessment of severe PH. Specific indications listed in Table 26.2. 2The degree of septal wall flattening in end-systole provides an estimate of RV systolic pressure (RVSP). 3Diastolic LV EI is a reflective marker of RV volume overload and systolic EI reflects RV pressure overload. A pressure-loaded RV in cPH will deviate the septum in systole and reduce the perpendicular dimension, resulting in an end-systolic LV EI ≥ 1.0. 4Visual inspection of the Doppler flow envelope across the RV outflow tract has been shown to be a sensitive predictor of altered pulmonary hemodynamics in neonates with PH. The characteristic mid-systolic notch (the “flying W”) and its different patterns integrate all the indicators of pulmonary vascular load and RV function and have been used to detect a fall in the RV afterload during the early transitional period in healthy term infants. 5RV peformance FAC, TAPSE, tissue Doppler imaging, and deformation have all been validated in term and preterm infants with emerging reference patterns in health and disease states. 6LV perfomance systolic disease is pretty rare in this setting – diastolic failure appears to be the primary contributor. Although several surrogate quantitative echocardiography methods are available to evaluate and monitor RV afterload, it is usually desirable, whenever feasible, to indirectly calculate pulmonary arterial pressures. This may be achieved by interrogation of regurgitation jets across tricuspid or pulmonary valves or flow across a PDA or VSD, as available. Among these, by far the most commonly employed method in clinical practice is estimation of RV systolic pressure from the tricuspid regurgitation jet velocity (TRJV) using the modified Bernoulli equation (RVSP = 4 × [TRJV]2 + right atrial pressure).48 In addition, PVR can be assessed with the TRJV interrogation based on its relationship to velocity time integral along the RV outflow tract (TRJV:VTI),49 as well as dynamic compliance,50 and pulmonary artery capacitance.51 While these may add value to our understanding of the disease and degree of pulmonary bed vasoreactivity, their use in preterm neonates with cPH needs validation. A complete tricuspid regurgitation jet, however, is only present in ∼30% of cPH cases at 36 weeks PMA, limiting the clinical applicability of this measure. A complete pulmonary valve regurgitation jet is even less often available, but when present, it may be used to measure both mPAP and end-diastolic pulmonary pressure by peak regurgitant velocity and end-diastolic regurgitant velocity, respectively.52 Using TRJV to estimate RVSP, pulmonary artery diastolic pressure may also be estimated by the following equation: PADP = 0.49 × RVSP.53 Though, its utility in cPH in preterm neonates is unknown. Qualitatively, visual inspection of the intraventricular septal wall configuration, particularly degree of flattening in end-systole (parasternal short axis view) obtained at the level of papillary muscles, is used for assessment of presence and, semi-quantitatively, severity of cPH. This measure is the most frequently employed index in neonatal cPH studies to date, as it is easy and available in almost all patients, including sequential assessments. A flat interventricular septum at end-systole (“D-shaped” left ventricle) indicates RVSP to be between 50 and 100% of systolic blood pressure, while the septum being concave toward the left side (paradoxical septum motion) indicates RVSP to be greater than systolic blood pressure.54 While a perfectly round septum (“O-shaped” left ventricle) most certainly rules out cPH, and inter-rater reliability of this measure is very good at either extreme of disease, it remains a qualitative index with inferior reliability in moderate disease.55 The eccentricity index (EI) is a measure that quantifies the degree of septal flattening by calculation of the ratio between left ventricular anteroposterior and septolateral linear dimensions; a ratio >1.0 indicates raised RV pressures at the corresponding time in the cardiac cycle.56 While raised EI at end-diastole suggests RV volume overload (from ASD/PFO shunt), end-systolic EI reflects RV pressure loading.57 While exact diagnostic thresholds for cPH in neonates have not been well established, pilot studies indicate that an end-systolic EI ≥ 1.3 may reflect RVSP more than half-systemic and diminished RV systolic performance.58 However, EI may be a useful marker for longitudinal evaluation of disease severity and response to treatments. Qualitative presence of RV hypertrophy and dilation is also a strong indicator of cPH.56,59 Finally, it should be noted that in cases of severe hypertension, systolic EI may not be reliable. Pulse wave Doppler interrogation of the RV outflow tract and flow in the main pulmonary artery also provides supplementary indices, which are easy to obtain in the majority of patients and can be very useful for screening for pulmonary vascular disease and estimation of change over time or response to therapies.60 The presence of a mid- or late-systolic notch in the Doppler trace of RV outflow tract is indicative of raised PVR, validated against invasive measurement in adult patients.61 With regards to time periods on the Doppler trace of the main pulmonary artery, the pre-ejection period, referring to the time between the onset of ventricular depolarization and onset of ejection of blood across the pulmonary valve, is directly proportional to PAP.62 RV ejection time (RVET) is relative to stroke volume and has an inverse relationship to pulmonary artery compliance. Pulmonary artery acceleration time (PAAT), considered to be the most useful time-dependent index in cPH, is the time interval from the onset to the peak velocity of Doppler trace and is inversely related to PVR and systolic PAP.41 PAAT and its ratio with RVET have been validated against cardiac catheterization measures of PVR and cPH in children and adults,41 and changing patterns over time have been established for term and preterm infants.34,63,64 Although PAAT <70 ms at 36 weeks PMA in preterm neonates is suggested to reflect raised pulmonary arterial pressure,65 specific validated diagnostic thresholds for cPH, which correspond to risk of adverse outcomes, are not known. We find PAAT and its ratio to RVET to be highly useful in clinical practice and recommend sequential evaluations for preterm neonates being scanned for cPH and related follow-up. The intricate cardiopulmonary interactions and myocardial remodeling seen in cPH may result in varying degrees of systolic and diastolic RV dysfunction in the context of alterations in afterload. Morphological measures of RV performance provide diagnostic clues as to how the structure of the RV responds to increases in afterload. Assessment of RV performance may be characterized by three separate techniques (Figure 26.3): (i) change in cavity dimensions (e.g., RV fractional area change [FAC]); (ii) displacement and velocity of single point along the myocardial wall (e.g., tricuspid annular plane systolic excursion [TAPSE], tissue Doppler imaging, tissue Doppler velocities); and (iii) deformation of a segment of the wall (e.g., strain analysis).66 RV FAC reflects a change in cavity size and is a surrogate of RV ejection fraction.67 Although typically measured from the apical four-chamber view, evidence suggests that capturing the FAC from the apical three-chamber view accurately reflects the longitudinal shortening in systole from the RV infundibulum and is less influenced by interventricular septal motion.68 Recent studies of BPD-associated cPH reported lower values of FAC at 36 weeks PMA compared to asymptomatic preterm infants.67,69 Although RV FAC linearly increases with gestational age at birth and weight, normal values are considered to be >35% by 1 month of age.67,70 TAPSE measures the longitudinal distance (displacement) that the tricuspid valve annulus moves during systole and provides an estimate of longitudinal myocardial shortening and RV systolic performance. Although it is an easily reproducible measure that is not influenced by heart rate,71 TAPSE only reflects the displacement of the tricuspid annulus at a single point and does not account for the tethering effect of regional wall motion abnormalities and segmental RV dysfunction.72 Similar to FAC, TAPSE is a load-dependent measure,73 with established normative data at birth in preterm infants.72 Low TAPSE < 4 mm predicts the need for ECMO or death in term infants with acute PH.74 TAPSE increases linearly from early gestation to term equivalency; therefore these cut-off values cannot be extrapolated to preterm infants with cPH. In a prospective multicenter study TAPSE was decreased in preterm infants with cPH as early as 1 month of age compared to term infants and preterm infants without BPD or evidence of cPH.26 Emerging evidence suggests that TAPSE is decreased in some preterm infants with cPH at 36 weeks PMA,19,75 although specific values that predict adverse outcomes are not known.
Chapter 26: Chronic pulmonary hypertension
Introduction
Epidemiology and risk factors
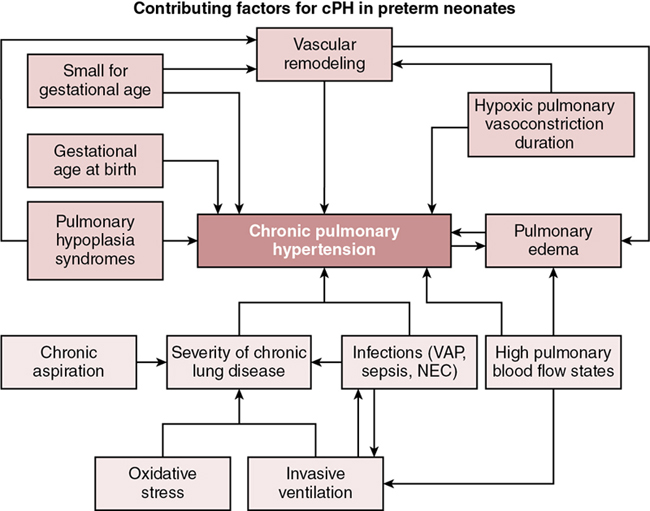
Neonatal right ventricle in cPH: Main determinant of symptoms and outcome
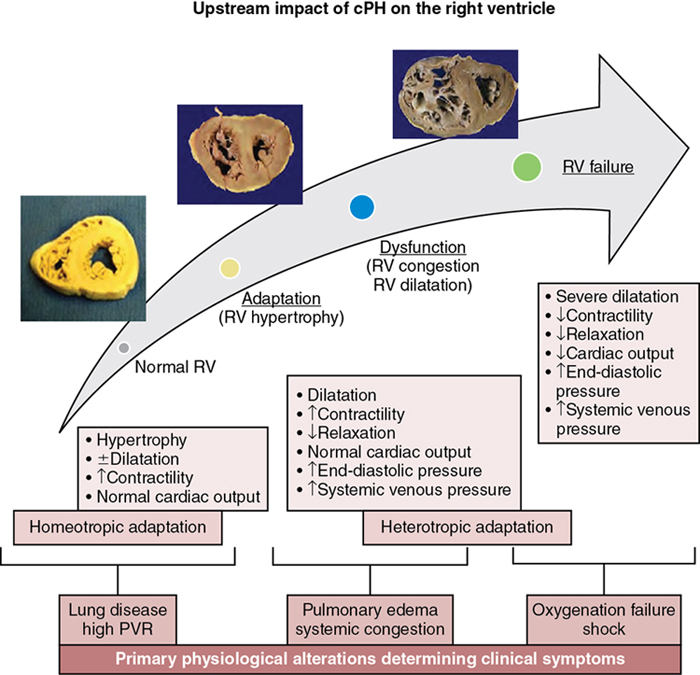
Clinical symptoms and signs
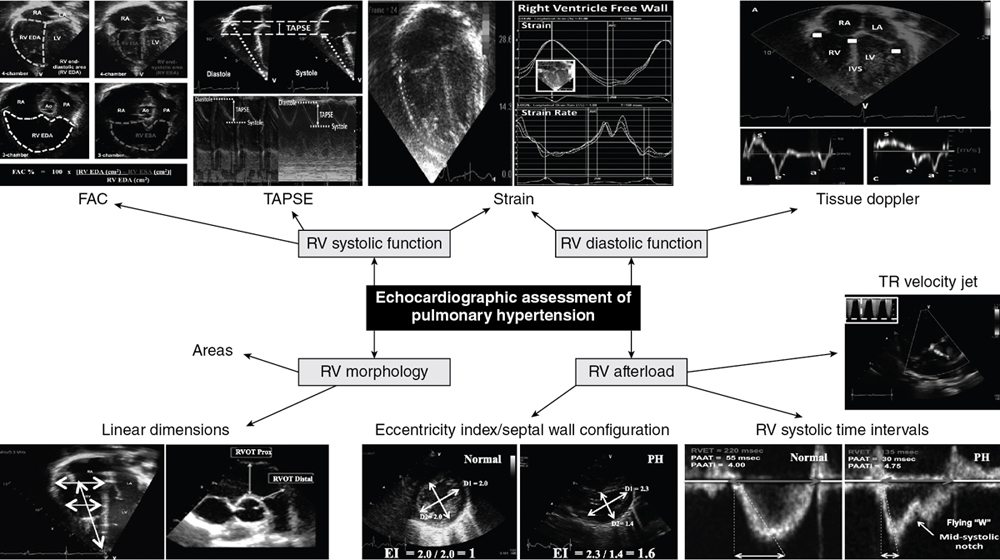
Diagnostic assessment for cPH in preterm neonates
Diagnostic Approach
Pulmonary Hypertension Assessment
I. Echocardiography
Clinical standard investigation for screening of patients with CLD, Establish initial diagnosis and severity of cPH. Utilize for longitudinal monitoring to detect disease progression and response to interventions.
II. Cardiac catheterization
Although gold-standard investigation, clinical utilization highly limited by risk of complications owing to patient size and stability. Currently, its use is reserved for select circumstances, such as (a) severity and underlying pathophysiology of PH not clear on echocardiography, (b) need to identify contributing cardiovascular co-morbidities and role of cardiac shunts, and (c) need to elucidate underlying pathophysiology and confirm vasoreactivity in cases of poor response or worsening with PH-targeted therapy.
III. Cardiac magnetic resonance
Provides a noninvasive option to assess right and left heart size and function, along with pulmonary and systemic flow mechanics and shunt fraction (phase contrast MRI), especially useful in patients that are not candidates for cardiac catheterization. Currently, availability is limited to specialized centers and often is cumbersome to organize and can only be used for sporadic assessment, even when available.
IV. Computerized tomography
Is particularly used as an aid for the evaluation of chronic lung disease, airway disease, pulmonary artery size, and identification of pulmonary vein stenosis.
Role of comprehensive echocardiography in cPH assessment in neonates
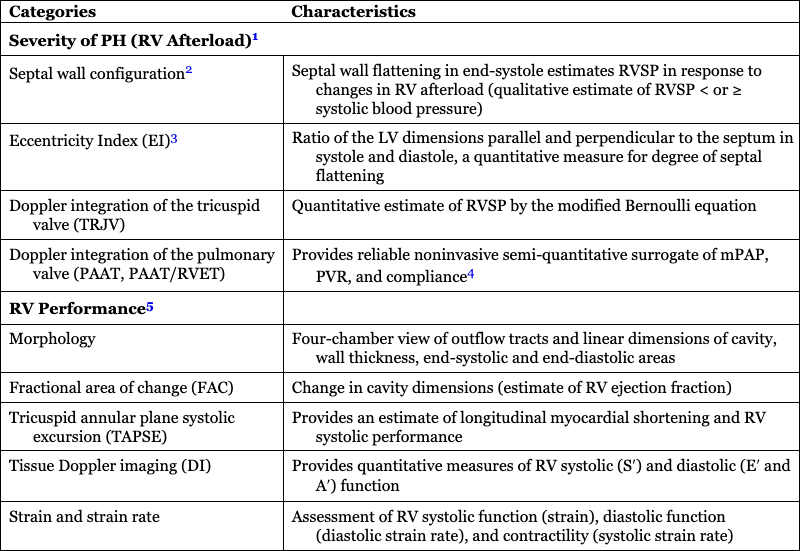
Categories
Characteristics
Severity of PH (RV Afterload)1
Septal wall configuration2
Septal wall flattening in end-systole estimates RVSP in response to changes in RV afterload (qualitative estimate of RVSP < or ≥ systolic blood pressure)
Eccentricity Index (EI)3
Ratio of the LV dimensions parallel and perpendicular to the septum in systole and diastole, a quantitative measure for degree of septal flattening
Doppler integration of the tricuspid valve (TRJV)
Quantitative estimate of RVSP by the modified Bernoulli equation
Doppler integration of the pulmonary valve (PAAT, PAAT/RVET)
Provides reliable noninvasive semi-quantitative surrogate of mPAP, PVR, and compliance4
RV Performance5
Morphology
Four-chamber view of outflow tracts and linear dimensions of cavity, wall thickness, end-systolic and end-diastolic areas
Fractional area of change (FAC)
Change in cavity dimensions (estimate of RV ejection fraction)
Tricuspid annular plane systolic excursion (TAPSE)
Provides an estimate of longitudinal myocardial shortening and RV systolic performance
Tissue Doppler imaging (DI)
Provides quantitative measures of RV systolic (S′) and diastolic (E′ and A′) function
Strain and strain rate
Assessment of RV systolic function (strain), diastolic function (diastolic strain rate), and contractility (systolic strain rate)
RV velocity time integral (VTI) and RV output
Estimate of RV stroke volume and, in the absence of a PDA, pulmonary blood flow
RV-PA coupling (TAPSE/PAAT and strain/PAAT)
Reliable estimate of invasive coupling hemodynamics (not validated in preterm neonates with cPH)
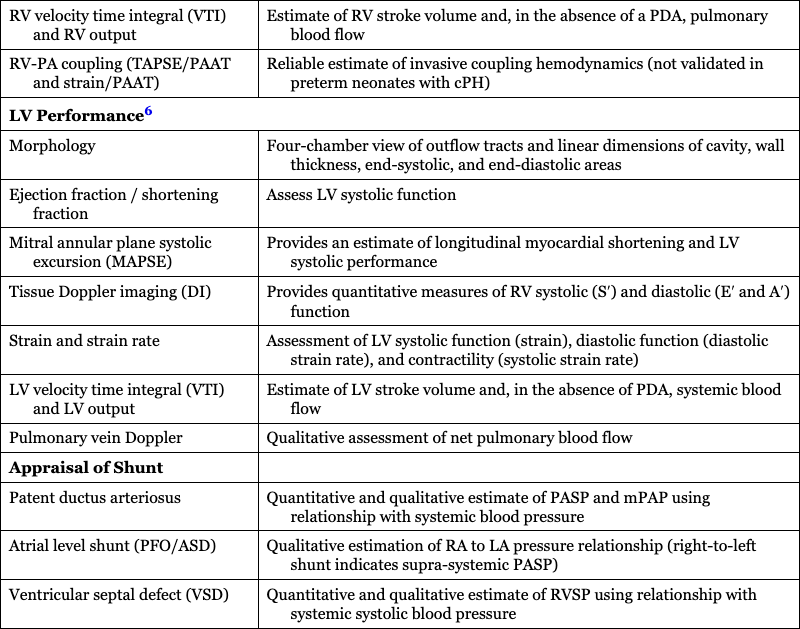
LV Performance6
Morphology
Four-chamber view of outflow tracts and linear dimensions of cavity, wall thickness, end-systolic, and end-diastolic areas
Ejection fraction / shortening fraction
Assess LV systolic function
Mitral annular plane systolic excursion (MAPSE)
Provides an estimate of longitudinal myocardial shortening and LV systolic performance
Tissue Doppler imaging (DI)
Provides quantitative measures of RV systolic (S′) and diastolic (E′ and A′) function
Strain and strain rate
Assessment of LV systolic function (strain), diastolic function (diastolic strain rate), and contractility (systolic strain rate)
LV velocity time integral (VTI) and LV output
Estimate of LV stroke volume and, in the absence of PDA, systemic blood flow
Pulmonary vein Doppler
Qualitative assessment of net pulmonary blood flow
Appraisal of Shunt
Patent ductus arteriosus
Quantitative and qualitative estimate of PASP and mPAP using relationship with systemic blood pressure
Atrial level shunt (PFO/ASD)
Qualitative estimation of RA to LA pressure relationship (right-to-left shunt indicates supra-systemic PASP)
Ventricular septal defect (VSD)
Quantitative and qualitative estimate of RVSP using relationship with systemic systolic blood pressure
RV afterload assessment and pulmonary hemodynamics
Assessment of RV performance
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Obgyn Key
Fastest Obstetric, Gynecology and Pediatric Insight Engine
