Chapter 44 Bleeding and Bruising
ETIOLOGY
What Platelet Problems Cause Bleeding?
Platelets may fail to function properly or may be decreased in number. Increased destruction and decreased production both result in a decrease in platelets (Table 44-1). Most disorders in both categories occur infrequently, but all must be considered carefully.
Table 44-1 Disorders Associated with Decreased Platelet Count
| Increased Destruction |
| Immune mediated (ITP, isoimmune) |
| Disseminated intravascular coagulation (DIC) |
| Sepsis |
| Hemolytic uremic syndrome (HUS) or TTP |
| Kasabach-Merritt syndrome (hemangiomas) |
| Hypersplenism |
| Decreased Production |
| Viral illness |
| Medications (prescription and OTC) |
| Leukemia or infiltrative malignancy |
| Chemotherapy drugs |
| Thrombocytopenia with absent radii |
| Amegakaryocytic thrombocytopenia |
| Wiskott-Aldrich syndrome |
| Aplastic anemia |
| Congenital marrow failure syndromes |
ITP, Idiopathic thrombocytopenic purpura (immune-mediated thrombocytopenia); OTC, over the counter; TTP, thrombotic thrombocytopenic purpura.
What Coagulation Problems Must I Consider?
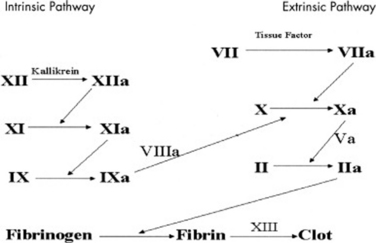
Anything that disrupts either the intrinsic or extrinsic coagulation pathway (Figure 44-1) will result in bruising and bleeding. Congenital deficiency of factors VIII and IX are commonly referred to as hemophilia A and B, respectively. These X-linked disorders are the most common congenital abnormalities of the fluid phase of coagulation.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


