Fig. 5.1
Molecular structure of bisphosphonates. The generic bisphosphonate structure (left) with two phosphonate groups binding to a central carbon atom. Modern bisphosphonates have a hydroxyl group and specific nitrogen-containing side chain (right) attached to the central carbon atom. (Black sphere: central carbon atom, orange spheres: phosphorus, red spheres: oxygen, grey spheres: hydrogen)
Pamidronate and zoledronate are the most widely used intravenous bisphosphonates in children with OI, with zoledronate being about 850 times more potent [4]. Zoledronate has a high affinity for bone and has a 100-fold bone concentration compared to plasma. Adult studies have shown that the effects of zoledronate are also longstanding with only a slight decline after 6 months [5].
The use of bisphosphonates has been well established in adult patients for treatment of osteoporosis, Paget disease of bone, myeloma, hypercalcaemia and bone metastases. Its use in children is relatively more recent but bisphosphonate therapy is now the mainstay of medical treatment for osteogenesis imperfecta (OI) [6].
Osteogenesis Imperfecta
Before discussing the role of bisphosphonates in OI it is important to understand the pathogenesis and heterogeneity of this condition. OI is characterised by increased bone fragility and decreased bone mass. There is significant variability in the clinical features and severity within OI, with presentation at any age from intrauterine life to adulthood. The extreme variability in OI results in part from genetic and biochemical heterogeneity. The diagnosis can be straightforward in the more severe cases or when there is a positive family history and the typical features of blue sclera, fracture and Wormian bones of the skull are present (Fig. 5.2). However, a definitive diagnosis can be more difficult on clinical grounds alone in milder cases that lack these features and it is not uncommon for mildly affected adults to only be identified when their affected child presents with fractures. Other features associated with OI include joint hypermobility, hearing impairment, basilar invagination and brittle teeth (dentinogenesis imperfecta).
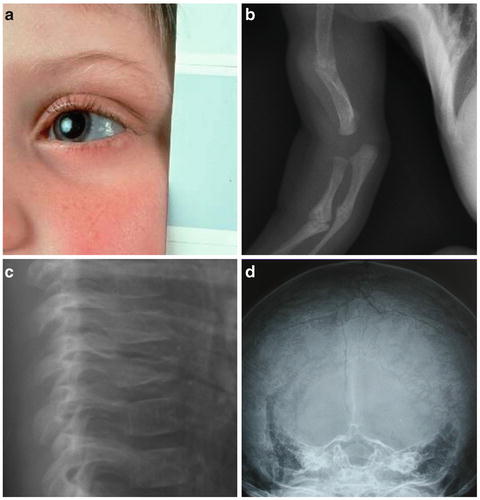
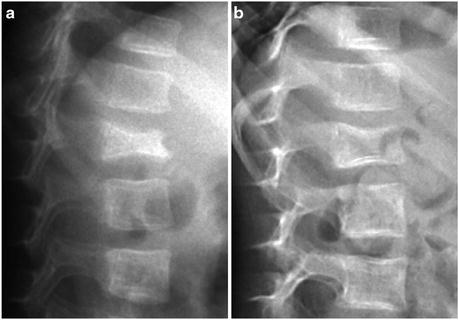
Fig. 5.2
Features of osteogenesis imperfecta. Blue sclerae (a), fractures ((b) multiple upper limb fractures, (c) vertebral compression fractures) and Wormian bones of the skull (d) are associated features of osteogenesis imperfecta
Various classification systems have been previously proposed to describe the heterogeneity of OI. In 1979, Sillence proposed a classification of OI types I–IV based on clinical findings with radiological subclassification [7]. Despite recent revisions of this classification to encompass the expanding molecular diagnosis, a variation of the original Sillence classification is still widely used in clinical practice (Table 5.1).
Table 5.1
Expanded Sillence classification illustrating five clinically distinct OI phenotypes
OI type | Phenotypic features | Inheritance |
|---|---|---|
OI type I | Osseous fragility (variable from none through moderately severe), distinctly blue sclerae (at all ages), presenile hearing loss (or family history of hearing loss) | AD |
OI type II | Perinatally lethal OI. Extremely severe osseous fragility, stillbirth or death in the newborn period. Rare long-term survival | AR and AD |
OI type III | Moderately severe to severe osseous fragility, normal sclerae (blue in infancy) ± severe deformity of long bones and spine. A range of clinical and radiographic phenotypes | AR and AD |
OI type IV | Osseous fragility with normal sclerae (blue in infancy) ± severe deformity of long bones and spine | AD |
OI type V | Osseous fragility with normal sclerae ± severe deformity + calcification in interosseous membranes ± hyperplastic callus | AD |
Type I OI is the most common type of OI and is distinctive because of blue sclerae and minimal bony deformity. Type I OI is classically due to reduction in the quantity of collagen type I protein due to a stop, frameshift or splice site mutation in either COL1A1 or COL1A2. As this leads to a quantitative defect, the phenotype of this group tends to be mild with patients attaining normal height and having minimal functional limitations. Recurrent long-bone or vertebral fractures can however result in significant disability in some. They can present as back pain, scoliosis or be asymptomatic and during growth there is good potential to respond to treatment. The incidence of hearing impairment increases during adult life.
Type II OI is associated with perinatal lethality. These infants are the most severely affected individuals with multiple fractures and bone deformities in utero and at birth. The sclerae of these children are often deep blue or grey but can be normal. Skeletal survey reveals bones that are short and broad with very low density. Respiratory insufficiency is the leading cause of mortality due to multiple rib fractures and pulmonary insufficiency. Mutations in these babies tend to be sporadic and typically involve a glycine substitution in COL1A1 or COL1A2.
Type III is the most severe form of OI in those who survive the neonatal period. They often have multiple long-bone deformities and fractures at birth, have blue or grey sclerae, severe dentinogenesis imperfecta and triangular facies. Over 50 % are wheelchair dependent at an early stage and have very short stature. These children have severe bone fragility throughout their childhood, which can in turn lead to limb and spinal deformities and result in respiratory compromise. Genetic mutations in these individuals are often due to a glycine substitution in COL1A1 or COL1A2 leading to a qualitative defect in the collagen protein. Mutations in other genes (see below) can lead to a very similar severe phenotype.
Type IV OI is characterised by osteoporosis leading to bone fragility without the typical features of the type I phenotype (i.e. blue sclerae and deafness). Fractures may present at any age and a majority of these patients have short stature. A small proportion experience a severe, progressive lower limb deformity rather than recurrent fractures. Dentinogenesis imperfecta is variable but when present is associated with a greater frequency of fractures. Inheritance is autosomal dominant with mutations in COL1A1 and COL1A2.
Type V OI was the first non-collagen OI type to be identified and constitutes about 4 % of the OI population. Mutations of the IFITM5 gene have been shown to cause this autosomal dominant form of OI [8]. Classically, patients have a distinctive phenotype with moderate-to-severe bone fragility although recent family studies have shown the phenotype is variable. The hallmark of this subtype is the presence of hypertrophic callus formation and early calcification of the interosseous membrane between the bones of the forearm, which limits pronation and supination. Radial head dislocation can be identified from a young age and predates interosseous membrane calcification. Scleral colour is normal. Upon histological examination, the lamellar organisation of the bone has an irregular mesh-like appearance, clearly distinct from the normal lamellar organisation seen in type I and IV OI.
A number of other subtypes of OI have been classified based on different genetic, phenotypic or histomorphometric features. While it is important to distinguish the genetic cause for genetic counselling, the most salient clinical feature regarding the role of bisphosphonate treatment is the severity of bone fragility. Type VI OI clinically resembles other forms of moderate-to-severe OI but it has a characteristic “fish-scale” pattern of bone lamellation on bone histology. Blue sclerae and dentinogenesis imperfecta are absent in type VI OI. Mutations in SerpinF1 have been shown to cause this subtype of OI [9] and there is evidence suggesting that response to bisphosphonate therapy, particularly gains in mobility scores and reductions in fracture incidence, is less than in other types of OI [10]. Type VII OI is a rare autosomal recessive condition that was described in the First Nations community in northern Quebec [11]. It is caused by mutations in CRTAP [12] and is associated with a moderate-to-severe phenotype involving fractures from birth, bluish sclerae, early lower limb deformity, coxa vara and osteopenia. Rhizomelia is a prominent clinical feature that distinguishes this form of OI.
Over 90 % of European individuals with OI have mutations in collagen type I genes (COL1A1 or COL1A2) [13]. In other populations such as Southern Asian or Samoan populations, evidence suggests that mutations in other genes may be more prevalent as causes of moderate-to-severe OI.
Collagen type I has a triple helical structure consisting of two α1 and one α2 chains. For the triple helix to fold correctly, every third amino acid residue must be a glycine with the remaining amino acids rich in proline and hydroxyproline [13]. The α-chains are initially synthesised as pro-α-chains with polypeptide extensions at either end. The carboxy propeptide extension is essential for pro-α-chain association prior to triple helix assembly, which occurs in a carboxy to amino direction. Using COL1A1 and COL1A2 mutation analysis alone, it is difficult to make genotype–phenotype correlations in OI. In general, however, mutations resulting only in a quantitative defect in collagen type I production result in a milder phenotype than those leading to a qualitative defect [14]. A number of additional genes have been identified that play a role in type I collagen trafficking and these have been implicated in autosomal recessive types of OI; FKBP10 [15], SerpinH1 (coding for HSP47) [16], SerpinF1 [9] and SP7/0SX (Osterix) [17]. Mutations in these genes result in moderate-to-severe OI and have subtle radiological features that may help to distinguish them from OI caused by COL1 mutations.
Disruption of collagen results in a disorder of the mineral phase of OI bone. Human OI bone has a higher average material density than normal bone, and the murine model of moderate-to-severe OI (OIM mouse) has smaller and less well-aligned mineral crystals than the wild-type mouse [18]. It is the combination of the organic and inorganic abnormalities of OI bone that alters its biomechanical properties and makes it brittle. Histomorphometric studies of OI bone have shown a decrease in core width, cortical thickness and trabecular number and thickness [19]. Individual osteoblasts produce a reduced amount of bone in OI, but due to their increased number, the bone formation rate is increased. Osteoclastic activity is also increased so this does not lead to a net gain in bone mass. Together these findings indicate a high turnover state with minimal net gain in bone mass. The increase in bone turnover is reflected in increased serum and urinary levels of markers of bone resorption (deoxypyridinoline and N-telopeptide) and bone formation (alkaline phosphatase and osteocalcin). The reduction in core width seen on trans-iliac bone biopsies translates into thinner long bones with a reduced polar moment of inertia, further increasing the propensity to fracture.
Bisphosphonates in OI
The aim of treatment in OI is to reduce fracture frequency, maximise mobility and improve functional outcomes [20]. The use of bisphosphonates to treat OI was first described in a case report in 1987 [21]. A 12-year-old girl with OI was treated with oral pamidronate. The dosing regime comprised 250 mg daily for 2 months alternating with 2 months of abstinence for a total duration of 1 year. She showed a 33 % increase in lumbar spine bone mineral content by dual photon densitometry but still went on to sustain at least two low-trauma fractures within the following year. The first systematic assessment of bisphosphonates in OI was undertaken over a decade later when the effects of cyclic intravenous pamidronate were investigated in 30 children with severe OI [22].
Intravenous pamidronate in children with OI has been reported to decrease bone pain, enhance well-being, improve mobility and muscle strength, reduce fracture incidence, increase long bone cortical thickness, increase vertebral size with vertebral reshaping and increase bone mass and bone mineral density [22, 23]. In an attempt to prevent growth disturbance and spine and limb deformity, cyclical intravenous pamidronate has also been used in babies and infants with OI [24]. The treatment response in the younger children was more pronounced than in the older cohort and there was also an improvement in the time taken to attain motor milestones [25]. Histomorphometry has provided valuable insight into the actions of pamidronate in children with OI [26]. The major bone effects of pamidronate were to increase cortical thickness and trabecular number. Trabecular thickness was not enhanced. Bone turnover was significantly reduced with a decrease in both bone resorption and formation below that of age-matched normal controls. There was also an increase in residual calcified cartilage within the bone. In adults with OI, pamidronate has been shown to increase spine and hip areal bone mineral density and decrease fracture rates [27]. The results in adults have not been as marked as those in children, suggesting that bisphosphonate therapy should be instigated during childhood to obtain maximal benefit.
A prospective randomised trial of 23 children with OI was carried out to assess the dosage, efficacy and safety of zoledronate compared to pamidronate [28]. This study showed a similar response in terms of improvements in serial bone density and quality of life. An increased frequency of fracture was seen in the zoledronate treatment group, which may have been due to inclusion of some children with more severe OI. Zoledronate has the benefits of being able to be administered more rapidly and of having a longer dosing interval than pamidronate, both of which may prove advantageous to patients and health care facilities.
As mentioned above, the primary aim of bisphosphonate treatment in OI is to reduce fracture frequency in order to maximise mobility. The best possible outcome is the ability to walk and bisphosphonates have revolutionised this aspect of patient management, particularly in those with moderate-to-severe disease (Figs. 5.3 and 5.4). Cyclical intravenous bisphosphonate therapy improves linear growth and bone structure via improvements in bone shape and biomechanics. Orthopaedic intervention in combination with bisphosphonate therapy has resulted in improvements in patient mobility [29].
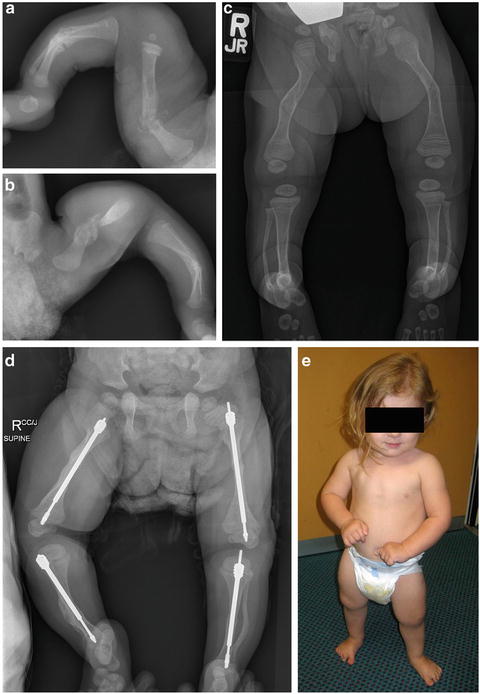
Fig. 5.3
Improvements in appendicular skeleton following bisphosphonate treatment. Multiple fractures in the neonatal period of (a) right leg and (b) left leg in a child with osteogenesis imperfecta. Cyclical intravenous bisphosphonate therapy aided fracture prevention and allowed linear growth (c). Note sclerotic bisphosphonate treatment lines, particularly in distal femur and proximal tibia. The bisphosphonate-treated bones were more amenable to orthopaedic intervention allowing the insertion of Fassier-Duval telescopic rods (d) resulting in the ability to mobilise independently (e)