Anti-tumor necrosis factor
Adalimumab
Monoclonal antibody
Crohn disease
Approved phase III
Ulcerative colitis
Certolizumab pegol
Pegylated antibody fragment
Crohn disease
Approved phase III
Golimumab
Monoclonal antibody
Ulcerative colitis
Phase III
CDP571
Monoclonal antibody
Crohn disease
Phase III—failed
Etanercept
Soluble receptor fusion protein
Crohn disease
Phase II—failed
Onercept
Soluble receptor
Crohn disease
Phase II—failed
Anti-adhesion
Natalizumab
Monoclonal antibody
Crohn disease
Approved phase III
VEDOLIZUMAB
Monoclonal antibody
Crohn disease
Phase III
Ulcerative colitis
Phase III
Alicaforsen
Antisense
Crohn disease
Phase III—failed
Ulcerative colitis
Phase II
Anti-interleukin 12/23
ABT-874 (J695)
Monoclonal antibody
Crohn disease
Phase II
CNTO 1275
Monoclonal antibody
Crohn disease
Phase II
Apilimod mesylate (STA-5326)
Small molecule
Crohn disease
Phase II
Anti-interleukin-2 receptor (anti-CD25)
Daclizumab
Monoclonal antibody
Ulcerative colitis
Phase II—failed
Basiliximab
Monoclonal antibody
Ulcerative colitis
Phase II—failed
Miscellaneous
Sargramostim
Recombinant protein
Crohn disease
Phase III—failed
Filgrastim
Recombinant protein
Crohn disease
Phase II
Interleukin 10
Recombinant protein
Crohn disease
Phase III—failed
Fontolizumab (anti-interferon γ)
Monoclonal antibody
Crohn disease
Phase II
Visilizumab (anti-CD3)
Monoclonal antibody
Crohn disease
Phase II
Ulcerative colitis
Phase III—failed
RDP58
Peptide
Crohn disease
Phase II—failed
Ulcerative colitis
Phase II—failed
Abatacept (anti-CTLA-4)
Receptor fusion protein
Crohn disease
Phase III
Ulcerative colitis
Phase III
Antagonist to chemokine receptor 9
CCX282-B
Small molecule
Crohn disease
Phase II
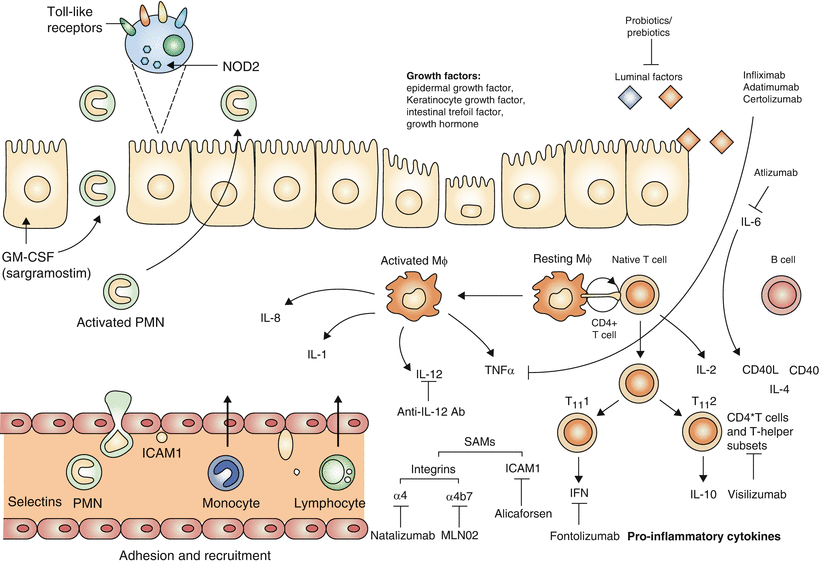
Fig. 34.1
Novel therapeutic targets in inflammatory bowel disease (IBD). Potential therapies in IBD encompass interventions at a variety of pathways in the inflammatory cascade. These include altering luminal factors, enhancing intestinal repair, augmenting the intestinal innate immune barrier function, inhibiting cell adhesion, and blocking cytokine activity. GM-CSF, granulocyte–macrophage colony-stimulating factor; ICAM-1, intercellular adhesion molecule 1; IFN, interferon; IL, interleukin; Mø, macrophage cell; PMN, peripheral blood mononuclear cell; SAM, selective adhesion molecule. Reprinted with permission from: Korzenik JR, Podolsky DK. Evolving knowledge and therapy of inflammatory bowel disease. Nature Reviews Drug Discovery 2006;5:197–209
Anti-Tumor Necrosis Factor
The anti-tumor necrosis factor (TNF)-α agents other than infliximab that have been evaluated for the treatment of Crohn disease include adalimumab (D2E7), certolizumab pegol (CDP871), CDP571, golimumab, etanercept, and onercept.
Adalimumab
Adalimumab is a fully human IgG1 monoclonal antibody directed against TNF-α. Adalimumab binds to TNF-α and blocks its interaction with the p55 and p75 cell surface TNF receptors, it fixes complement, mediates antibody-dependent cytotoxicity, and induces T-cell apoptosis [1, 2]. It is administered subcutaneously, and has a half-life of 12–14 days. Controlled trials have shown that adalimumab is effective for the treatment of rheumatoid arthritis [3–10], plaque psoriasis, psoriatic arthritis [11], juvenile idiopathic arthritis, and ankylosing spondylitis [12], and have led to regulatory approval for these indications. In addition, four placebo-controlled trials have demonstrated that adalimumab is effective for the induction and maintenance of remission in adult patients with Crohn disease and based on these studies it received regulatory approval in the United States for this indication in 2007 [13–16].
In the CLASSIC I trial, 299 patients with moderate-to-severe Crohn disease who were previously naïve to TNF therapy were randomized to receive one of the four subcutaneous induction loading dose regimens at weeks 0 and 2: adalimumab 40 mg/20 mg, 80 mg/40 mg, or 160 mg/80 mg or placebo. The 40 mg/20 mg loading dose was anticipated to be subtherapeutic. The adalimumab loading doses of 80 mg/40 mg and 160 mg/80 mg were chosen to achieve adalimumab serum concentrations at week 4 that would be seen at steady state (12–16 weeks) with adalimumab 40 mg every other week and 40 mg weekly, respectively. The rates of remission (Crohn disease activity index [CDAI] < 150 points) at week 4 in the adalimumab 40 mg/20 mg, 80 mg/40 mg, and 160 mg/80 mg groups were 18%, 24%, and 36%, respectively, and 12% in the placebo group [13]. These results demonstrated that 160 mg/80 mg (equivalent to 40 mg weekly dosing) was the optimal induction regimen. Two hundred seventy six of 299 patients enrolled in the CLASSIC I trial continued in the CLASSIC II maintenance trial. All patients received adalimumab 40 mg at week 0 (week 4 of CLASSIC I) and week 2. Fifty five patients who remained in remission at week 0 and 4 were re-randomized to receive adalimumab 40 mg every other week, adalimumab 40 mg every week, or placebo. The rates of remission at week 56 in the placebo, adalimumab 40 mg every other week, and adalimumab every week were 39%, 69%, and 83%, respectively [14]. The remaining 221 patients who did not maintain remission at week 0 and/or week 4 received open-label adalimumab 40 mg every other week, with dose escalation to weekly doses of 40 mg for flare or nonresponse. The rates of response-70 (decrease from baseline in the CDAI ≥ 70 points) and remission at week 56 were 69% and 44%, respectively. Forty-six percent of patients required dose escalation to adalimumab 40 mg weekly [14]. Concomitant immunomodulator therapy did not significantly affect the rates of response-70 and remission [14, 17].
The CHARM trial examined maintenance of remission in patients with moderate-to-severe Crohn disease who responded to induction therapy with adalimumab, including those who had previously been treated with infliximab (these patients were a mixture of patients who responded to infliximab and stopped therapy, and patients who responded to infliximab and lost response or became intolerant). Eight hundred fifty-four patients received open-label adalimumab 80 mg at week 0, then 40 mg at week 2. At week 4, 58% of patients achieved response-70, and were randomized to receive adalimumab 40 mg weekly, 40 mg every other week, or placebo up to week 56 [15]. Those who had a flare or nonresponse could switch to open-label 40 mg every other week after week 12. At week 26, the rates of response-70 in the placebo, adalimumab 40 mg every other week, and adalimumab every week groups were 28%, 54%, and 56%, respectively [15]. The rates of remission in the placebo, adalimumab 40 mg every other week, and adalimumab every week groups were 17%, 40%, and 47%, respectively [15]. The rates of response-70 at week 56 in the placebo, adalimumab 40 mg every other week, and adalimumab every week groups were 18%, 43%, and 49%, respectively [15]. The rates of remission at week 56 in the placebo, adalimumab 40 mg every other week, and adalimumab every week were 12%, 36%, and 41%, respectively [15]. There was no statistical significance between the rates of response-70 and remission in the groups randomized to receive adalimumab weekly or every other week. The rates of remission were similar regardless of whether patients received prior infliximab or not, and whether they received concomitant immunosuppressive therapy (azathioprine, 6-mercaptopurine, methotrexate) or not [18]. Of 117 patients with fistulas, complete healing was achieved in 33% of those who received adalimumab, compared to 13% who received placebo [15, 19]. Adalimumab was also steroid-sparing [15, 20]. These results demonstrated that 40 mg every other week was the optimal maintenance regimen, with dose escalation to 40 mg weekly for sustained nonresponse or loss of response.
There are currently three approved anti-TNF agents used for the treatment of patients with Crohn disease; adalimumab, certolizumab pegol, and infliximab. Each agent is different in structure and does not cross react with others. If patients lose response or have adverse events occur that preclude further use of infliximab it was uncertain if the use of adalimumab was safe until the GAIN (Gauging Adalimumab Efficacy in Infliximab Nonresponders) trial was performed. The GAIN trial examined maintenance of remission in patients with moderate-to-severe Crohn disease who had previously responded to infliximab and then lost response or became intolerant [16]. Three hundred eleven patients were randomized to receive adalimumab (n = 155) 160 mg at week 0 and 80 mg at week 2, or placebo (n = 156) [16]. Patients received subcutaneous injections of adalimumab, 160 mg at week 0, and 80 mg at week 2, at the same schedule as placebo. Investigators assessed disease activity at week 0 (pretreatment) and weeks 1, 2, and 4. At 4 weeks, the rates of clinical responses and remission for adalimumab and placebo significantly differed: 52% vs. 34% (≥70-point decrease in the CDAI score); 38% vs. 25% (≥100-point decrease in CDAI score); and 21% vs. 7% remission (CDAI score ≤ 150 points). The effect of adalimumab on fistulas was not different from that of patients who were treated with placebo. The clinical improvement with adalimumab was not dependent upon the reason for stopping infliximab therapy and was unaffected by the absence or presence of antibodies to infliximab (ATIs) [16].
In patients with rheumatoid arthritis, the rate of formation of anti-adalimumab antibodies is 5% (1% for patients receiving concomitant therapy with methotrexate and 12% for patients not receiving methotrexate) [21]. The incidence of anti-adalimumab antibodies in the CLASSIC II trial was 3% [14].
Recently, several trials have also been performed assessing the efficacy of adalimumab in the treatment of patients with ulcerative colitis. The first study assessed the efficacy and safety of adalimumab for the induction of clinical remission in anti-TNF naıve patients with moderately to severely active ulcerative colitis [22]. This 8-week multicenter study was conducted in a randomized, double-blind placebo-controlled fashion [22]. Ambulatory adult patients were enrolled who had a Mayo score ≥6 points and endoscopic subscore of ≥2 points despite treatment with corticosteroids and/or immunosuppressants. Initially, 186 patients were randomized to receive either Adalimumab 160 mg at week 0, 80 mg at week 2, 40 mg at weeks 4 and 6, or placebo [22]. Subsequently, as a consequence of the demands of the European regulatory authorities, the protocol was amended with the addition of a second induction group (Adalimumab 80 mg at week 0, 40 mg at weeks 2, 4, and 6) [22]. The primary efficacy endpoint in this study was clinical remission (defined as a Mayo score ≤ 2 with no individual subscore > 1) at week 8 [22]. This endpoint was evaluated in 390 patients who were randomized to adalimumab 160 mg initially then 80 mg at week 2 or adalimumab 80 mg initially then 40 mg at week 2, or placebo [22]. Clinical remission was achieved at week 8 in 18.5% of patients in the adalimumab 160/80 group (p = 0.031 vs. placebo), 10.0% in the adalimumab 80/40 group (p = 0.833 vs. placebo), and 9.2% in the placebo group [22]. Serious adverse events occurred in 7.6%, 3.8%, and 4.0% of patients in the placebo, adalimumab 80/40, and adalimumab 160/80 groups, respectively [22]. The authors of this study thus concluded that adalimumab 160/80 was safe and effective for induction of clinical remission in ambulatory patients with moderately to severely active ulcerative colitis failing treatment with corticosteroids and/or immunosuppressants [22].
Another study evaluated induction and maintenance in adult patients with active ulcerative colitis [23]. The study was a randomized, double-blind, placebo-controlled trial which evaluated the efficacy of adalimumab in induction and maintenance of clinical remission in 494 patients with moderate-to-severe ulcerative colitis who received concurrent treatment with oral corticosteroids or immunosuppressants [23]. Eligible patients were outpatients who had moderately to severely active Ulcerative Colitis for at least 3 months with a Mayo score of 6–12 points (endoscopy subscore of at least 2), despite concurrent therapy with steroids and/or azathioprine or 6-mercaptopurine [23]. Patients were stratified based upon their prior exposure to TNF-antagonists (either had or had not been previously treated with anti-TNF therapy) and they were randomly assigned to groups given adalimumab 160 mg at week 0, 80 mg at week 2, and then 40 mg every other week or placebo [23]. The primary study end points were remission at weeks 8 and 52. At the period of 8 weeks the rates of clinical remission were 16.5% in those patients on adalimumab and 9.3% on placebo (p = 0.019) with rates at 52 weeks being 17.3% and 8.5% (p = 0.004), respectively [23]. In anti-TNF-naïve patients, the rates of remission at week 8 were 21.3% on adalimumab and 11% on placebo (p = 0.017); and at week 52 rates were 22% and 12.4% (p = 0.029), respectively [23]. In patients who had received prior anti-TNF agents, remission at week 8 was 9.2% on adalimumab and 6.9% on placebo (p = 0.559); and at week 52 remission rates were 10.2% and 3%, respectively (p = 0.039) [23].
Pediatric Data
A recent multicenter international pediatric trial evaluated the efficacy of adalimumab for induction and maintenance for 1 year in 188 children aged 6–17 years (mean age 13.5) [24]. Patients had moderately to severely active Crohn disease (defined by PCDAI > 30) despite concurrent treatment with oral corticosteroid and/or an immunomodulator use [24]. There were 105 anti-TNF naïve patients and 83 had previously been treated with and responded to infliximab, but had experienced secondary loss of response or intolerance. All patients in this study received medical therapy in open-label fashion. They received a 2-dose induction regimen of adalimumab based on body weight (160 mg initially and then 2 weeks later 80 mg if their body weight was greater than or equal to 40 kg). If their body weight was less than 40 kg they received 80 mg initially and 40 mg 2 weeks later. Overall a total of 82% of the overall patients (155/188) responded (defined as a drop in PCDAI of ≥15 points) at week 4. All patients in the trial (regardless of week 4 initial response status) were then randomized (following stratification according to prior anti-TNF exposure) to maintenance adalimumab every 2 weeks at either high dose adalimumab (40 mg every other week if body weight ≥ 40 kg; 20 mg eow if body weight < 40 kg) or low dose adalimumab (20 mg every other week if body weight ≥ 40 kg; 10 mg every other week if body weight < 40 kg).
The primary outcome for this study was clinical remission (defined as a PCDAI ≤ 10) at week 26. Over half (57%) of the anti-TNF naïve children treated with open-label induction and then high dose every other week maintenance were in clinical remission at week 26, and 45% achieved remission at week 52. These remission rates were superior to those achieved with low dose every other week maintenance. As is expected the remission rates in children who had been previously treated with infliximab (but lost response or became intolerant to infliximab) were lower with a 20% remission rate at week 26 and an 18% rate at week 52 even for those patients who received high every other week dosing.
Pregnancy
Published data on the use of adalimumab in pregnant women is limited to case reports. The initial report was of a woman with Crohn disease who received infliximab during one pregnancy and adalimumab throughout a second pregnancy. Both children were born full term. The second child was delivered via Cesareans section due to perianal disease. Both were developmentally normal at 6 months of age [25]. Subsequently, ten additional pregnancies in patients with Crohn disease have been reported [26–29].
Safety
The most common adverse effect in the Phase II and Phase III trials in patients with Crohn disease was localized injection reactions. In patients with Crohn disease treated with adalimumab, drug-induced lupus, demyelination, lymphoma, and serious and opportunistic infections have all been reported [30]. The serious infections include pneumonia, tuberculosis, nocardiosis, and coccidioidomycosis. In the controlled portions of the 32 global Adalimumab clinical trials in adult patients with Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis, Crohn Disease, and Psoriatic Arthritis, the rate of serious infections was 4.7 per 100 patient-years in 6,694 adalimumab-treated patients vs. a rate of 2.7 per 100 patient-years in 3,749 control-treated patients. Serious infections observed included pneumonia, septic arthritis, prosthetic and post-surgical infections, erysipelas, cellulitis, diverticulitis, and pyelonephritis [21].
Two patients with Crohn disease in the CHARM trial developed tuberculosis [15]. A recent meta-analysis of serious infection and malignancy rates in patient with rheumatoid arthritis treated with adalimumab and infliximab may have over-estimated the infection rates due to flaws in methodology [31]. In post-marketing surveillance, the rate of serious infections in adalimumab-treated patients was 4.1 per 100 patient-years, which was similar to the general rheumatoid-arthritis population. The rate of tuberculosis in post-marketing surveillance after initiation of TB screening is 0.27 per 100 patient-years. The rate of reported cases of lupus-like syndrome is 0.1 per 100 patient-years. The rate of demyelinating disorders (including multiple sclerosis and Guillian-Barré syndrome) was 0.08 per 100 patient-years. Analysis of the incidence of lymphoma in adalimumab clinical trials, in comparison with that of the normal population in the SEER database, resulted in an SIR of 3.19 (95% CI, 1.78–5.26). The types of lymphomas include Hodgkin’s disease, B cell lymphoma, T-cell lymphoma, central nervous system lymphoma, and mucosa associated lymphoid tissue lymphoma; none of which are predominant [32]. In the rheumatoid arthritis controlled trials, 12% of patients treated with adalimumab and 7% of the placebo-treated patients that had negative baseline ANA titers developed positive titers at week 24 [21].
Two patients of the 3,046 patients treated with adalimumab developed clinical signs suggestive of new-onset lupus-like syndrome. The patients improved after cessation of therapy. No patients developed lupus nephritis or central nervous system involvement [21].
Certolizumab Pegol (CDP870)
Certolizumab is a humanized TNF-α Fab monoclonal antibody fragment linked to polyethylene glycol that is administered subcutaneously. In vitro, certolizumab pegol has a high affinity for TNF-α, it is devoid of the Fc portion of the antibody and does not induce complement activation or antibody-dependent cellular cytotoxicity, and it does not induce apoptosis in T cells or macrophages [33–35].
In a placebo-controlled, phase II study, patients with Crohn disease with moderate to severely active Crohn disease were randomized to receive subcutaneous certolizumab pegol 100, 200, or 400 mg or placebo at weeks 0, 4, and 8. The response rates in the certolizumab pegol 400 mg group were significantly greater than in the placebo group at 2, 4, 6, and 10 weeks, but not at 12 weeks (which was the primary endpoint of the study) [36]. The best separation of certolizumab pegol treated patients from placebo-treated patients occurred in a subgroup of patients with elevated concentrations of C-reactive protein (≥10 mg/L) at baseline. Another small phase II study of intravenous certolizumab pegol also failed to achieve the primary endpoint of the study [37].
The PRECiSE trials are phase III trials of certolizumab in patients with active Crohn disease. PRECiSE 1 is a randomized, double-blinded induction with a late efficacy measurement (26 weeks) [38]. Six hundred sixty patients were stratified according to their baseline CRP concentration (<10 mg/L, ≥10 mg/L) and use of immunosuppressants and then randomized to receive certolizumab 400 mg subcutaneously or placebo at weeks 0, 2, 4, then every 4 weeks up to 24 weeks. At week 4, in the overall patient population (both CRP < 10 mg/L and ≥10 mg/L), there was a higher rate of clinical remission (19.5% vs. 11.3%) and clinical response (defined as a ≥100 point decrease in the CDAI score from baseline) (28.7% vs. 21.8%) in the certolizumab pegol group compared to placebo. At week 26, the rate of clinical response in the certolizumab pegol group was significantly higher than the placebo group (23.1% vs. 16%, respectively), while the rate of clinical remission trended in favor of certolizumab pegol. The co-primary endpoints for the study, clinical response at week 6 and 26, were also significantly greater in the certolizumab pegol group. Median CRP concentrations were significantly reduced in the certolizumab pegol group at weeks 6 and 26. PRECiSE 2 is a randomized, double-blinded maintenance trial after initial open-label induction [39]. In PRECiSE 2, 668 patients with active Crohn disease received open-label certolizumab 400 mg subcutaneously at weeks 0, 2, and 4 weeks. Those who had clinical response at week 6 (n = 428, 64%) were then randomized to receive double-blinded certolizumab 400 mg or placebo every 4 weeks up to week 24. Remission rates at week 26 (after 20 weeks of blinded therapy) were 47.9% in the certolizumab group compared to 28.6% in the placebo group [39].
A subsequent trial was performed to further evaluate the efficacy of induction therapy with certolizumab pegol in patients with active Crohn disease [40]. This study was a placebo-controlled trial that was conducted in 439 adult patients who had moderate-to-severe Crohn disease who had not been previously treated with anti-TNF therapy. Patients in the trial received either certolizumab pegol at a dose of 400 mg subcutaneously at weeks 0, 2 and 4 or placebo in a double-blind fashion [40]. The primary endpoint of this trial was clinical remission at week 6. The rates of clinical remission in patients receiving certolizumab pegol compared to placebo at 2 weeks, 4 weeks, and 6 weeks, respectively were 23% vs. 16% (p = 0.033), 27% vs. 19% (p = 0 0.063), 32% vs. 25% (p = 0.174), and 32% vs. 25% (p = 0.174) [40]. Patients who had C-reactive protein ≥ 5 mg/L had higher rates of clinical remission than placebo. In this study there were significant differences in patients who had increased concentrations of C-reactive protein when the study began between remission rates in patients who received certolizumab pegol vs. those who received placebo [40]. The authors thus stress that future clinical trials should emphasize the treatment of patients who have objective evidence of inflammation in addition to symptoms of active disease. Additional data from the PRECiSE 4 study demonstrated that administration of a single additional dose of certolizumab pegol to patients who initially respond and are on continuous maintenance therapy is a successful method to regain remission [41]. In addition, reinduction (certolizumab pegol 400 mg) subcutaneously given at week 0, 2, and 4 is an effective means to recapture remission in patients who relapsed after drug interruption [41].
Pediatric Data
There are no published clinical trial data on the use of certolizumab pegol in children or adolescents. Certolizumab pegol is currently, however, being studied in children and adolescents with CD in a phase II clinical trial (NCT00899678) [42]. This study is 62-weeks duration and is estimated that it will enroll 160 subjects aged 6–17 years. Certolizumab pegol dosing for patients 20–40 kg and over 40 kg will receive an induction dose of 200 mg or 400 mg, respectively, at weeks 0, 2, and 4. After this induction regimen, the patients will be randomized to a high- or low-dose regimen: in the high dose arm, patients over 40 kg will receive certolizumab pegol 400 mg and patients 20–40 kg will receive 200 mg every 4 weeks. In the low dose group those patients who are over 40 kg and patients 20–40 kg will receive certolizumab pegol at doses of 200 mg and 100 mg, respectively, every 4 weeks. The primary outcome of this phase II study will be the proportion of patients in clinical remission (defined as a Pediatric Crohn Disease Activity Index (PCDAI) score ≤ 10) at week 62 [42].
Safety
The most common side effect reported in both patients with Crohn disease and rheumatoid arthritis is headache [36–39, 43, 44]. Nasopharyngitis was also common in the Crohn disease trials [36–39, 43]. Serious infection occurred in 2–3% of patients treated with certolizumab pegol (including one patient who developed tuberculosis) and ≤1% of patients treated with placebo [38, 39]. Malignancy occurred in two patients treated with certolizumab pegol (metastatic lung cancer and adenocarcinoma of the rectum) and two patients treated with placebo (cervical cancer and non-Hodgkins lymphoma) [38, 39]. The two deaths that occurred during the PRECiSE trials (fentanyl overdose and myocardial infarction in a patient with metastatic lung cancer) were not thought to be related to certolizumab [38, 39]. No cases of lupus have been reported in any trial. In the PRECiSE trials the proportion of antinuclear antibodies was ≤8.3%, with ≤1.4% anti-dsDNA [38, 39].
CDP571
CDP571 is a humanized monoclonal IgG4 antibody to TNF which does not fix complement or mediate antibody-dependent cytotoxicity.
A small phase IIa study suggested that CDP571 might be efficacious in patients with active Crohn disease [45]. In a subsequent phase II study 169 patients with active Crohn disease were randomized to receive an initial dose of CDP571 10 or 20 mg/kg, or placebo [46]. At week 2, clinical response was significantly greater in the 10 mg/kg group as compared to placebo, but not the 20 mg/kg group [46]. Patients were then retreated with CDP571 10 mg/kg or placebo either every 8 or 12 weeks over a 24-week period. Significant differences in clinical response or remission over placebo were not achieved. A phase III study compared treatment of 396 patients with active Crohn disease randomized to CDP571 10 mg/kg or placebo every 8 weeks to week 24 [47]. CDP571 showed efficacy in achieving a statistically significant clinical response at week 2, but not at week 28. Post hoc analysis showed that patients with a baseline CRP ≥ 10 mg/L demonstrated higher clinical response over placebo at both week 2 (49.5% vs. 15.5%) and week 28 (28.7% vs. 12.1%) [47]. One study of 82 patients with active Crohn disease showed steroid-sparing effects of CDP571 over placebo at week 16 after an induction dose of 20 mg/kg at week 0, followed by 10 mg/kg dose at week 8 [48]. However, another study of 271 patients comparing CDP571 10 mg/kg or placebo administered every 8 week to week 32 failed to show steroid-sparing benefit in active Crohn disease [49].
Pediatric Data
An open-label study was conducted in 20 pediatric patients with active Crohn disease [50]. All were given a single dose of intravenous CDP571 10 mg/kg and were followed for 12 weeks. At week 2, 1 month, 65% had responded to treatment.
Etanercept
Etanercept is a fully human recombinant fusion protein that comprises an IgG1 Fc antibody fragment and two soluble p75 receptors to TNF. Etanercept does not fix complement, mediate antibody dependent cellular cytotoxicity, or induce T-cell apoptosis [51]. It is administered subcutaneously. Controlled trials have shown that etanercept is effective for the treatment of psoriasis [52–55], psoriatic arthritis [56, 57], rheumatoid arthritis [58, 59], ankylosing spondylitis [60–64], and juvenile rheumatoid arthritis [65], and have led to regulatory approval for these indications.
A controlled trial at a dose effective for rheumatoid arthritis (25 mg subcutaneously twice weekly) over an 8-week period failed to show efficacy in patients with moderate-to-severe Crohn disease [66].
Pediatric Data and Safety
There are no published data on the safety or efficacy of etanercept in children or adolescents with ulcerative colitis or Crohn disease. Given the lack of efficacy of etanercept for Crohn disease in adults, the dosing of etanercept in children and adolescents with juvenile rheumatoid arthritis and the safety profile of etanercept will not be discussed here.
Onercept
Onercept is a recombinant human soluble p55 TNF receptor. Although an open-labeled pilot study of two dosages of onercept in patients with active Crohn disease showed a dose–response effect in rates of remission and response [67], a subsequent phase II placebo-controlled trial failed to demonstrate efficacy over placebo [68].
Anti-Adhesion Molecules
Natalizumab
Natalizumab is a humanized IgG4 monoclonal antibody against the adhesion molecule α4 integrin, which is involved in migration of leukocytes across the endothelium, and is up-regulated in sites of inflamed endothelium. It is administered intravenously every 4 weeks. The efficacy of natalizumab for the treatment of multiple sclerosis has been demonstrated in controlled trials [69–71]. In November 2004 natalizumab was approved by the FDA for the treatment of multiple sclerosis with subsequent withdrawal from the market in February 2005, and then reintroduction with certain restrictions for the treatment of multiple sclerosis in September 2006.
Six randomized double-blind placebo-controlled trials assessed the efficacy of patients with Crohn disease, whereas only one uncontrolled pilot study has been conducted in patients with ulcerative colitis.
An initial phase IIa trial comprises 30 patients with active Crohn disease who received randomly allocated single infusion of either natalizumab 3 mg/kg or placebo and had CDAI evaluated 2 weeks after infusion [72]. There was a significant decrease in baseline CDAI score in patients treated with natalizumab at week 2 (mean drop 45 points, p = 0.02) whilst no significant decrease was observed in placebo arm (mean drop 11 points, p = 0.2). It demonstrated a higher rate of clinical remission at week 2 in patients given natalizumab 3 mg/kg compared to placebo (39% vs. 8%, p = 0.1) [72]. There was no significant difference between natalizumab and placebo in achieving remission at week 2 (39% vs. 8%, p = 0.1). This pilot trial did not show any significant superiority of natalizumab over placebo in treating patients with Crohn disease.
The second trial compared natalizumab (single infusion of 3 mg/kg, two infusions of 3 mg/kg or two infusions of 6 mg/kg) vs. placebo in 248 patients with moderate-to-severe Crohn disease [73]. The primary endpoint (remission, CDAI < 150 points at week 6) rates were 29% for single infusion of natalizumab (p = 0.757), 44% for two infusions of natalizumab 3 mg/kg (p = 0.030) and 31% for two infusions of natalizumab 6 mg/kg (p = 0.533) vs. 27% in placebo arm. Only two doses of natalizumab 3 mg/kg had statistically significant superiority over placebo in achieving clinical remission.
Three phase III trials have been conducted in Crohn disease. In Efficacy of Natalizumab as Active Crohn Therapy (ENACT-1), 905 patients with moderate-to-severe Crohn disease were randomly assigned to receive induction therapy at weeks 0, 4, and 8 with either natalizumab 300 mg or placebo [74]. The primary endpoint in the induction trial was clinical response defined as at least 70-point decrease in baseline CDAI score at week 10 and it was achieved in 56% and 49% of natalizumab and placebo recipients, respectively (p = 0.05) [74]. In ENACT-2, 339 patients who had a response to natalizumab in induction ENACT-1 trial at both week 10 and 12 were randomly reassigned to receive 300 mg of natalizumab or placebo every 4 weeks from week 12 through week 56 [74]. The primary endpoint in ENACT-2 trial was a sustained response through week 36. Patients with at least 70-point increase in CDAI score after week 12, with an absolute CDAI score of at least 220 or needed therapeutic intervention after week 12 were considered losing response. Rates of sustained response at week 36 were 61% in patients receiving maintenance treatment with natalizumab and 28% in those receiving placebo maintenance (p < 0.001). It was demonstrated that maintenance treatment with natalizumab is significantly superior to placebo in patients who responded to induction treatment. Concomitant immunosuppressants did not improve the rates of clinical remission or response [75]. Patients who maintained remission on natalizumab over 12 months in the ENACT-2 trial were enrolled into a subsequent phase III, open-label, 2-year open-label extension trial designed to assess long-term efficacy and safety of natalizuma [76]. This open-label trial comprises 146 patients who received 12 natalizumab infusions over 12 months. The proportion of patients who maintained remission after 6 (week 24) and 12 (week 48) additional infusions of natalizumab was 89% and 84%, respectively. This open-label extension trial supported data from ENACT-2 trial that natalizumab maintains remission over additional 12 months in patients with sustained remission on natalizumab in the preceding 12-months.
In the ENCORE trial, 509 patients with moderate-to-severe Crohn disease with elevated C-reactive protein concentrations at baseline were randomized to receive natalizumab 300 mg or placebo at weeks 0, 4, and 8 [77]. Natalizumab was significantly superior over placebo in inducing remission at week 8 that was sustained through week 12 (primary endpoint defined as at least 70-point decrease in CDAI score) with respective proportions of patients of 48% vs. 32% (p < 0.001). In addition natalizumab was also significantly superior over placebo in achieving additional efficacy endpoint, namely clinical remission at week 8 sustained through week 12 (CDAI < 150) that was observed in 26% of natalizumab-treated patients and 16% of placebo recipients (p = 0.002).
Finally, Sands et al. performed a placebo-controlled trial in which 79 patients with active Crohn disease during ongoing treatment with infliximab 5 mg/kg every 8 weeks for at least 10 weeks before initiation of randomization were randomly assigned to receive three intravenous infusions of either natalizumab 300 mg or placebo every 4 weeks while continuing their initial infliximab regimen during duration of the trial [78]. At week 6, patients treated with natalizumab plus infliximab experienced mean decrease in their CDAI score of 37.7 points while those treated with placebo plus infliximab experienced small increase in CDAI score of a mean of 3.5 points (p = 0.084). A trend towards greater efficacy of combined treatment with natalizumab and infliximab over infliximab alone was shown in patients with active Crohn disease not responding to infliximab therapy.
Gordon et al. published results of one small open-label study of ten patients with active ulcerative colitis who were treated with a single infusion of natalizumab 3 mg/kg [79]. All patients had their disease activity evaluated using Powell–Tuck score 2 weeks after infusion. Treatment with natalizumab resulted in significant decrease in median disease activity score from 10 at baseline to 6 at 2 weeks post-infusion (p = 0.004). It was suggested that future randomized, placebo-controlled trials are warranted to further assess the efficacy of natalizumab in ulcerative colitis.
Pediatric Data
There was only one open-label study conducted in 38 pediatric patients (ages 12–17 years) with active Crohn disease that assessed the efficacy of natalizumab in a pediatric population [80]. Among 38 enrolled patients 31 of them received three intravenous infusions of natalizumab 3 mg/kg at weeks 0, 4, and 8. Disease activity was measured using Pediatric Crohn Disease Activity Index [PCDAI] at baseline and then every 2 weeks through week 12. There was a significant decrease observed in PCDAI score from baseline at every time point (p < 0.001) with the greatest decrease observed at week 10 with 55% of patients achieving clinical response (>15-point decrease from baseline) and 29% of patients achieving clinical remission (PCDAI < 10). These promising findings however need to be validated in large randomized controlled trials.
Safety
In one study in patients with Crohn disease, 7% of patients given one or two induction doses of natalizumab (at weeks 0 and 4) had formed anti-natalizumab antibodies at 12 weeks [73]. Patients in the ENACT-2 trial who received concomitant immunosuppressants did not develop persistent anti-natalizumab antibodies, compared to 7.5% of patients who received natalizumab alone [74]. In patients with multiple sclerosis, the rate of formation of anti-natalizumab antibodies was 9%, with persistence in 6% (antibodies detected ≥2 more than 42 days apart) [81].
The largest ENACT-1 (n = 905) and ENACT-2 (n = 339) trials of natalizumab observed that serious adverse events occurred in similar proportion of patients in both trials (7% in natalizumab and placebo arms in induction ENACT-1 trial and 8% in natalizumab arm and 10% in placebo arm in maintenance trial) [76]. However, one patient died (three doses of natalizumab combined with azathioprine during ENACT-1, placebo with azathioprine during ENACT-2, and five doses of natalizumab alone after completion of ENACT-2 trial) from progressive multifocal leukoencephalopathy (PML), associated with the JC virus was observed [76]. The other large induction trial ENCORE on 509 patients with adverse events observed similar proportion of adverse events between natalizumab (85%) and placebo (82%) arms without any deaths [77]. The most common adverse events that were observed in at least 10% among either treatment arms were headache, nausea, abdominal pain, nasopharyngitis, dizziness, fatigue, and exacerbation of Crohn disease. There was a significant greater proportion of patients in natalizumab group vs. placebo that experienced nasopharyngitis (11% vs. 6%, p < 0.05), headache (29% vs. 21%, p < 0.05), and hypersensitivity reaction (4% vs. 0.8%, p < 0.05). On the other hand, exacerbation of Crohn disease was observed in greater proportion of placebo-treated patients when compared to natalizumab (13% vs. 7%, p < 0.05). Antibodies to natalizumab were detected in 9.5 of 241 tested patients treated with natalizumab and in 0.8% of 236 tested placebo recipients (p < 0.05).
A placebo-controlled trial by Sands et al. assessed primarily safety of concurrent therapy with natalizumab in 79 patients with Crohn disease already receiving infliximab [78]. The observed incidence of adverse events was similar in the treatment groups (natalizumab plus infliximab vs. infliximab plus placebo). The most frequent adverse events in both groups were headache, Crohn disease exacerbation, nausea, and nasopharyngitis. No one experienced a hypersensitivity-like reaction to natalizumab, whilst four patients (5%) experienced such reactions to infliximab. The development of antibodies to natalizumab was reported in 4% of patients whereas antibodies to infliximab were detected in 14% of patients.
Data from pediatric open-label study showed that the most common adverse events were headache (26%), pyrexia (21%), and exacerbation of Crohn disease (24%) [80]. Anti-natalizumab antibodies were detected in 8% of patients.
Clinical trials and marketing of natalizumab were suspended in February 2005 after two patients with multiple sclerosis treated with natalizumab and interferon beta-1A developed PML from reactivation of the latent human Jacob Creutzfeldt polyoma virus [82, 83]. A third patient treated with natalizumab and prior exposure to azathioprine was reclassified from malignant astrocytoma to PML [84]. An independent adjudication committee performed a safety evaluation in all patients who had recently been treated with natalizumab in clinical trials [85]. Evaluation consisted of a referral to a neurologist, brain magnetic resonance imaging, and polymerase chain reaction analysis of cerebral spinal fluid and serum for JC virus. Of 3,826 initial patients enrolled in clinical trials of natalizumab, safety evaluation included 87% (1,275), 91% (2,248), and 92% (296) of patients with Crohn disease, multiple sclerosis, and rheumatoid arthritis patients. No additional cases of PML were identified [85]. The median duration of treatment for all patients was 17.9 months, while that of patients with Crohn disease was 7 months. The absolute risk of developing PML during treatment with natalizumab was 1:1,000 (0.1%) with 95% confidence intervals of 1:200–1:2,800 [85]. The FDA reapproved natalizumab for multiple sclerosis in September 2006, with the requirement of mandatory participation in a risk management and registry program called the TOUCH program [81]. Natalizumab is currently under regulatory review for the treatment indication of Crohn disease.
Vedolizumab
Vedolizumab (also known as MLN-002 and MLN-02) is a recombinant IgG1-humanized monoclonal antibody against the adhesion molecule α4β7 integrin that is administered intravenously every 4 weeks. Deletion of the Fc receptor recognition and binding sites prevents complement fixation and subsequent release of cytokine.
At the time of writing this chapter there has been one published phase II study form that assessed vedolizumab in 185 patients with active Crohn diseas [86]. Patients were randomly assigned to either vedolizumab (0.5 mg/kg, n = 62 or 2.0 mg/kg, n = 65) or placebo (n = 58) on days 1 and 29 [86]. The primary endpoint was clinical response defined as at least 70-point decrease in the CDAI score on day 57 when compared to baseline [86]. There was no significant difference in the proportion of patients achieving the primary endpoint between vedolizumab given at dose 0.5 mg/kg (49%, p = 0.36) or 2.0 mg/kg (53%, p = 0.14) and placebo (41%). There was a statistically significant difference in clinical remission (CDAI ≤ 150) rates at day 57 (secondary endpoint) between high-dose vedolizumab recipients and placebo (37% vs. 21%, p ≤ 0.05). Likewise, high-dose vedolizumab was found to be significantly superior to placebo in achieving another secondary endpoint, enhanced clinical response (at least 100 point decrease in CDAI on day 57 when compared to baseline) with respective response rates of 47% and 31% (p ≤ 0.05). It was suggested that there is a dose-dependent efficacy of vedolizumab on achieving clinical remission in patients with ulcerative colitis.
There were two studies published that assessed vedolizumab in patients with ulcerative colitis. Data from a small phase IIa randomized double-blind placebo-controlled trial published only in an abstract form suggested that vedolizumab is safe and well-tolerated in the doses up to 2.0 mg/kg in patients with active ulcerative colitis [87]. Subsequently Feagan et al. published the results of a phase II study of vedolizumab in 181 patients with moderately severe active ulcerative colitis in which patients were randomly assigned to receive either vedolizumab (0.5 mg/kg or 2 mg/kg) or placebo on day 1 and day 29 [88]. The primary endpoint was clinical remission at week 6 that was defined as an ulcerative colitis clinical score of 0 or 1 and a modified Baron score of 0 or 1 with no evidence of rectal bleeding. At week 6, the rates of clinical remission for the vedolizumab at 0.5 mg/kg (33%, p = 0.02), 2 mg/kg (32%, p = 0.02) were significantly greater when compared to placebo (14%). Overall patients receiving active drug were more than twofold more likely to achieve remission than those receiving placebo (RR = 2.25, 95%; CI, 1.17–4.36) [88, 89]. Treatment with vedolizumab was also efficacious in achieving secondary endpoints at week 6, namely clinical response (at least 3 point reduction in the ulcerative colitis clinical score) and endoscopic remission (a modified Baron score of 0) when compared to placebo. Patients receiving vedolizumab were nearly twofold more likely than placebo recipients to achieve clinical response at week 6 (RR = 1.78, 95%; CI, 1.22–2.60) [88, 89]. The endoscopic remission rate at week 6 was 28% in the vedolizumab group at 0.5 mg/kg, 12% in the vedolizumab group at 2 mg/kg, and 8% in the placebo group (p = 0.007) [88]. However, when patients treated with vedolizumab were analyzed together the difference in endoscopic remission at week 6 was not statistically significant showing trend favoring vedolizumab (20% vs. 8%; p = 0.05; RR = 2.46, 95%; CI, 0.98–6.15) [88, 89].
Pediatric Data
At the time of writing this chapter there are no published data on the use of vedolizumab in children or adolescents with IBD.
Safety
There were no significant differences observed in frequency of adverse events between vedolizumab and placebo in the phase II studies of vedolizumab for Crohn disease or ulcerative colitis [86, 88]. Among patients with Crohn disease 92% of MLN0002-treated and 86% of placebo-treated patients experienced at least one adverse event and 13% of vedolizumab-treated and 17% of placebo-treated patients experienced at least one serious adverse events [86]. The most common serious adverse event was exacerbation of Crohn disease that was observed in 8% and 9% of vedolizumab and placebo recipients, respectively. Clinically significant (titers > 1:125) human anti-human antibody (HAHA) levels were observed on day 57 in 12% and 34% of patients in the 2.0- and 0.5-mg/kg vedolizumab treatment arms, respectively. Increased titers of HAHA were associated with lower remission rates when compared with patients who had low or absent HAHA titers (data not shown).
Data from trial in patients with ulcerative colitis showed the nonsignificantly increased relative risk of serious adverse events of 1.60 (95% CI, 0.67–3.83) in patients treated with vedolizumab vs. placebo [89]. Feagan et al. distinguished three notable adverse events that were observed in patients treated with vedolizumab: an infusion reaction (high HAHA titers) with hives and angioedema (n = 1), a primary cytomegalovirus infection (n = 1), and postoperative pneumonia after spine surgery (n = 1) [88]. The development of HAHA was reported in 44% of patients treated with vedolizumab and high HAHA titers (>1:125) were found in 38% and 11% of patients receiving low dose and high dose of vedolizumab, respectively. Of note, clinical remission rates were greater in patients with low or absent HAHA levels (42%) while in patients with high HAHA titers clinical remission rates of 14% were comparable to those observed in placebo recipients (12%).
AJM 300
AJM 300 is an orally active small molecule with antagonistic properties to α4-integrin. The only trial published in an abstract from a randomized trial involving 71 patients with active Crohn disease. This study compared oral treatment with either AJM 300 (40 mg tid, 120 mg tid, or 240 mg tid) or placebo for 8 weeks [90]. The primary endpoint was the decrease of CDAI score from baseline to final evaluation at week 4 or later while the secondary efficacy endpoint was clinical response (≥70 point decrease in CDAI). There was no significant difference in clinical response was observed between active treatment and placebo arms. Among patients with high CDAI at baseline a significant decrease from baseline CDAI score (mean decrease 41.5 points, p = 0.0485) was observed in those treated with AJM 300 at the dose of 120 mg tid and mean 41.6 point decrease from baseline CDAI in those treated with AJM 300 at the dose of 240 mg tid (p-value not reported). In addition, patients treated with AJM at the dose of 240 mg tid had significant twofold decrease in C-reactive protein from baseline over 8 weeks (p = 0.0220). The investigators suggested that AJM 300 at dose 120 and 240 mg tid showed clinical efficacy in treating patients with active Crohn disease [90].
Pediatric Data
At the time of writing this chapter there are no published data on the use of AJM 300 in children or adolescents with IBD.
Safety
AJM 300 was tolerated well with incidence of adverse events that was not dose-dependent (0.0%, 23.5%, and 22.2% for AJM 300 40 mg, 120 mg, and 240 mg treated patients, respectively, vs. 16.7% for placebo-treated patients, p-value not reported) [90].
Alicaforsen (ISIS 2302)
Alicaforsen (ISIS 2302) is a 20 base phosphorothioate oligodeoxy-nucleotide that hybridizes to a sequence in the 3′ untranslated region of intercellular adhesion molecule 1 (ICAM-1) mRNA [91]. The translated oligonucleotide-RNA serves as a substrate for the nuclease RNase-H, an ubiquitous intracellular endoribonuclease that recognizes DNA:RNA heteroduplexes as substrate for selective RNA hydrolysis. This results in reduction of ICAM-1 RNA expression and protein levels. Intravenous, subcutaneous, and rectal enema formulations have been studied in patients with Crohn disease or ulcerative colitis.
There have been three randomized placebo-controlled trials that assessed the efficacy of alicaforsen administered intravenously [92–94] and one randomized placebo-controlled trial that evaluated the efficacy of this agent administered subcutaneously [95] in patients with active Crohn disease.
A phase IIa double-blind randomized placebo-controlled trial of 20 patients with active Crohn disease suggested the efficacy of intravenously administered alicaforsen [92]. Patients were randomly assigned to be treated with 13 infusions of either alicaforsen (0.5, 1, or 2 mg/kg, n = 15) or placebo (n = 5) over the period of 26 days with subsequent 6 month follow-up [92]. The rates of clinical remission (CDAI < 150) at the end of treatment were 47% and 20% in active drug and placebo arms, respectively (p-value not reported) [92]. ISIS 2302 showed corticosteroid sparing effect with significantly lower dose of corticosteroids over time when compared to placebo (p = 0.0001). Data from subsequent dose ranging pharmacokinetic trial of high-dose alicaforsen administered intravenously at the dose of 300 or 350 mg three times a week for 4 weeks in 22 patients with active Crohn disease demonstrated that 41% of patients achieved clinical remission indicating that this agent might be efficacious in treating Crohn disease. Unfortunately, large randomized placebo-controlled trials with intravenous alicaforsen did not support these preliminary findings.
In the subsequent large clinical trial that comprises 299 patients with active steroid-dependent (prednisone 10–40 mg) Crohn disease patients were randomly assigned to intravenous treatment three times a week with either ISIS 2302 (2 mg/kg) or placebo for 2 or 4 weeks and the regimen was then repeated after 1 month without treatment [93]. The corticosteroid-free remission (CDAI < 150) at week 14 (primary endpoint) was comparable between combined ISIS 2302 and placebo arms (20.2% vs. 18.8%, p-value not reported). On the other hand, a significantly greater proportion of patients receiving ISIS 2302 than placebo had successful corticosteroids withdrawal at week 14 (78% vs. 64%; p = 0.032). According to pharmacodynamic analysis statistically significant results for clinical remission, improvement in CDAI, and quality of life based on IBD questionnaire were observed in the highest area under the curve subgroup of ISIS 2302 arm when compared to placebo. Finally, data from two double-masked, placebo-controlled trials of patients with Crohn disease who received intravenous treatment with either alicaforsen (n = 221) or placebo (n = 110) three times a week for 4 weeks did not show any benefit of alicaforsen over placebo in achieving clinical remission at week 12 with respective remission rates of 33.9 and 34.5% (p = 0.89) [94].
Subcutaneous administration of alicaforsen also did not demonstrate any superiority over placebo in achieving clinical remission in patients with Crohn disease. Schreiber et al. randomized 75 patients with corticosteroid-refractory Crohn disease to subcutaneous treatment with either ISIS 2302 or placebo [95]. The primary endpoint, corticosteroid-free remission at week 14 (CDAI < 150) was observed in 3.3% of ISIS-2302-treated and 0% of placebo-treated patients. On the other hand, there was a trend towards efficacy of ISIS 2302 in achieving one of the secondary endpoints, namely corticosteroid-free remission at week 26 (13.3% vs. 6.7%, p-value not reported). Similarly, a greater proportion of patients receiving active drug when compared to placebo achieved a corticosteroid dose <10 mg/day at week 14 (48.3% vs. 33.3%) and week 26 (55.0% vs. 40.0%) and a prednisone equivalent dose of 0 mg at week 26 (23.3% vs. 6.7%, respectively).
There have been three randomized placebo-controlled trials assessing the efficacy of alicaforsen enemas in patients with active left-side ulcerative colitis [96–98].
Van Deventer et al. performed a randomized placebo-controlled trial of alicaforsen enema in 40 patients with mild to moderately active distal ulcerative colitis who received 60 mL of alicaforsen enema (0.1, 0.5, 2, or 4 mg/mL) or placebo once daily for 28 consecutive days. There was observed a significant dose-dependent reduction in disease activity index in patients treated with active drug than placebo at day 29 that was observed for alicaforsen given at the highest dose 4 mg/mL (70% vs.28%, p = 0.004). After 3 months alicaforsen 2 and 4 mg/mL caused significant reduction in disease activity index when compared to placebo by 72% and 68%, respectively (vs. 11.5% for placebo, p = 0.016 and 0.021, respectively). In the subsequent phase II dose ranging, double-blind, placebo-controlled study of alicaforsen enema (120 mg daily for 10 days, then every other day; 240 mg every other day; 240 mg daily for 10 days, then every other day; 240 mg daily) given daily for 6 weeks in 112 patients presenting with acute exacerbation of mild to moderate left-sided ulcerative colitis; there was no significant difference observed between active drug and placebo in reduction of disease activity index at week 6 [97]. However, a greater proportion of patients receiving alicaforsen 240 mg daily had prolonged clinical improvement at week 18 (51% vs. 18%) and week 30 (50% vs. 11%) when compared to placebo. Finally, Miner et al. compared two dose formulations of alicaforsen enema (120 or 240 mg) with 4 g mesalamine enema given for 6 weeks in 159 patients with mild to moderate left-sided ulcerative colitis [98]. There was no difference observed between treatment arms in reduction of disease activity index at week 6 with reduction in mean disease activity index when compared to baseline of 50% for the mesalamine arm and 40 and 41% for the 120 and 240 mg alicaforsen groups (p = 0.27 and 0.32, respectively). However, higher dose of alicaforsen enema was significantly more efficacious than mesalamine in achieving clinical remission at week 18 (20% vs. 6%, p = 0.03) [98].
An open-label study of alicaforsen enema given at daily dose of 240 mg for 6 weeks to 15 patients with active ulcerative colitis showed a 46% reduction in mean disease activity index and 33% rate of complete mucosal healing at the end of treatment [99]. In addition, alicaforsen concentrations were greater in mucosal colonic tissue biopsies than those observed in plasma suggesting that alicaforsen enemas allow for achieving high local concentrations with little systemic exposure. Another open-label study of 12 patients with chronic pouchitis following an ileal pouch-anal anastomosis for ulcerative colitis showed that alicaforsen enemas given at dose of 240 mg daily for 6 weeks resulted in significant reduction in the mean pouchitis disease activity index from baseline value of 11.42 points to 6.83 points at 6 weeks (p = 0.001) [100].
Pediatric Data
At the time of writing this chapter there are no published data on the use of alicaforsen in children or adolescents with IBD.
Safety
Data from large trial of 331 patients treated with intravenous alicaforsen or placebo showed that the only adverse events that occurred in greater proportion of patients treated with alicaforsen were symptoms related to infusion reactions such as fever (22.6% vs. 14.7%, p-not significant), chills (14% vs. 1.8%, p = 0.0005), and myalgia (5.4% vs. 0.92%) [94]. Data from the second largest trial of 299 patients with Crohn disease receiving alicaforsen or placebo intravenously showed that the only adverse events that occurred in significantly greater proportion of patients treated with active drug than placebo were infusion reactions described as transient facial flushing or a feeling of warmth during infusion (11.6% vs. 4%, p = 0.03) [93]. There was a significantly greater average transient aPTT increase without bleeding sequelae (8.66 s vs. 0.8 s, p < 0.0001) after alicaforsen than placebo infusion [93]. Safety analysis of alicaforsen administered subcutaneously in the largest trial of 75 patients determined that injection site reactions, headache, pain, fever, rash, arthritis, asthenia, and flu-like symptoms injection site reactions occurred in greater proportion of patients treated with active drug than placebo with injection site reactions demonstrating the largest difference (23.3% vs. 0%, p-value not reported) [95].
Anti-Interleukin 12/23
Interleukin-12 and interleukin-23 are key pro-inflammatory cytokines involved in type 1 helper T (Th1) cell response which is characterized by a marked accumulation of macrophages making interleukin-12, the major Th1-inducing factor, in Crohn disease mucosa [101, 102]. A recent genome wide association study demonstrated a highly significant association between Crohn disease and ulcerative colitis and the interleukin 23 receptor gene on chromosome 1p31, which encodes a subunit of the receptor for interleukin-23 [103]. It was also suggested that the production of both IL-12 and IL-23 is down-regulated in patients with Crohn disease but not with ulcerative colitis following administration of IL-12p40 monoclonal antibody [104].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


