(CPP = cerebral perfusion pressure; CVR = cerebral vascular resistance), where CPP is the difference between the mean arterial blood pressure (MAP) and the ICP as seen in the equation below.
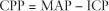
This pressure gradient ensures blood flow through the brain. Pathologic consequences occur if the CPP is either too low (ischemia) or too high (hyperoxia).
CBF and Metabolism
The gold standard for measurement of global CBF is the Kety-Schmidt technique (4). This relatively cumbersome technique requires cannulation of the jugular veins, and is not considered ideal for critically ill patients. Transcranial Doppler (TCD) sonography has been successfully used to measure blood flow velocities in the basal cerebral arteries. These measurements correlate well with changes in CBF (5,6) and the technique can be safely used at the bedside of critically ill patients.
CBF volume is significantly higher in children compared to adults. In the term neonate, blood flow volume averages ~70 mL/min (7), increasing 10-fold to ~700 mL/min by 3 years of age and peaking at ~900 mL by the age of 6.5 years before declining to ~700 mL at 15 years (8,9). The greatest increase occurs over the first 6 months of life (8). The increase in CBF volume occurs in parallel to increases in oxygen metabolic rate (10) and regional glucose metabolism (11) that reflect neuronal growth and maturation during infancy and early childhood (12). The coupling of blood flow to cerebral metabolism is an important concept, and has also been described during sleep (13) and general anesthesia (14).
Significant intracranial pathology often disrupts the mechanisms that regulate CBF, resulting in either excessive or insufficient blood flow to the brain. A recent study examining the relationship between CBF (by middle cerebral artery flow velocity, Vmca) and CPP in children following traumatic brain injury (TBI) found an inconsistent relationship between the two variables—79% of subjects with a CPP > 40 mmHg had a normal Vmca, while Vmca was decreased in 9% and increased in 12% (15).
Little data exists on the changes in metabolic rate or metabolism in the pediatric brain following trauma or other intracranial pathology; however, adult data suggests global cerebral metabolism is decreased following severe TBI (16). The association between hypotension, low CPP, and poor outcome in patients with severe TBI has been well documented (17,18). It has also been shown that CBF less than 10 to 12 mL/min/100 g causes irreversible global damage to neuronal tissue (19). Studies using microdialysis techniques to measure the metabolic requirements of injured versus noninjured tissue (20) demonstrate that injured brain may be more vulnerable to decreases in CBF and oxygen delivery (20).
Carbon Dioxide and Oxygen Vasoreactivity
Changes in the partial pressure of carbon dioxide rapidly alter CBF in a quasilinear fashion in the healthy adult. The vasodilatation associated with a 1 mmHg increase in PaCO2 results in a 2% to 4% increase in CBF (21). The reverse is also true—a decrease in PaCO2 causes marked vasoconstriction (22). These changes occur within seconds, making PaCO2 a potent cerebral vascular regulator (23). The effects are attenuated by hypotension (24), severe hypoxia (25), and barbiturates (26). Cerebral vasoreactivity is impaired during the initial acute phase following TBI (27,28), which predisposes the brain to either underperfusion or overperfusion. Adverse outcomes are reported in adult patients hyperventilated following TBI (29), and poor vascular response to changes in PaCO2 have been associated with negative neurologic outcomes in children (30). The most recent evidence-based TBI guidelines recommend that prophylactic hyperventilation should be avoided in adult (31) and pediatric (32) patients.
The effect the partial pressure of oxygen has on the cerebral vasculature is less dramatic, with hypoxia-induced vasodilatation (33) and hyperoxia-induced vasoconstriction (34,35) taking several minutes to occur.
Pressure Autoregulation
In healthy adults cerebral autoregulation maintains CBF constant over a range of MAP between 60 and 160 mmHg. The cerebral vasculature responds to changes in pressure by constricting and dilating to avoid rapid changes in blood flow. The effect is achieved by regulating arteriolar diameter so that a decrease in blood pressure results in vasodilatation to maintain blood flow, and a rise in blood pressure leads to vasoconstriction to protect cerebral vascular beds. Beyond the autoregulatory limits, CBF passively increases with an increase in CPP and ICP. The inverse is true with hypotension with associated risk of neural tissue ischemia. The response that cerebral vasculature has to changes in blood pressure is believed to invoke a myogenic effect (36), with smooth muscle tone being altered by changes in transmural pressure.
The other mechanism of pressure autoregulation is neurogenically mediated. Cerebral vessels have both sympathetic and parasympathetic innervation. The former are believed to have a significant effect on vasoconstriction through noradrenaline and neuropeptide Y (37). This has been demonstrated with an increase in CBF with stellate ganglion block (38) and trigeminal ganglion stimulation (39). The sympathetic innervation seems to shift the upper limit of autoregulation to the right, protecting the brain from episodes of hypertension.
Following moderate to severe TBI, cerebral pressure autoregulation is disrupted in both adult (40) and pediatric patients (41). There is evidence that this disruption is greater in cases of nonaccidental versus accidental injury (42). The outcome of patients with impaired pressure autoregulation remains poor in children (42) and adults (43).
Microcirculatory Changes
First elaborated in 1896, the Starling equation essentially states that the net flux of fluid into or out of capillaries (Jv) in the tissues depends on the balance between the hydrostatic pressure within the capillary (Pc) and interstitium (Pi), the oncotic pressure within the capillary (πc) and interstitium (πi), the filtration coefficient of the capillary membrane (or how leaky the capillary membrane is to fluid) (Kf), and the reflection coefficient of the capillary membrane (how leaky it is—or is not—to particular molecules) (σ). This relationship can be expressed by the formula:
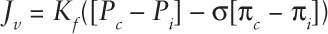
This concept is relevant when discussing ICP management in a number of areas, in particular, hyperosmolar therapy as well as the basis of what has become known as “the Lund concept” of approaching ICP management (44) (discussed in more detail below).
■ INDICATIONS FOR ICP MONITORING
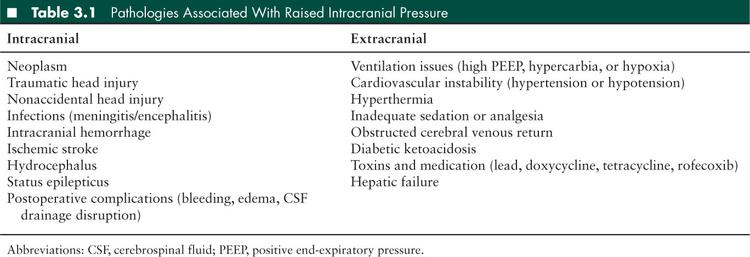
The clinical features of headache, nausea, or vomiting and decreased level of consciousness should alert the clinician to the possibility of intracranial hypertension. Pathologies that predispose to an increase in ICP can be divided into intracranial or extracranial (see Table 3.1). Fundoscopy and neuroimaging aid in the diagnosis, but elevated ICP may occur in the absence of either papilledema (45) or abnormal neuroimaging (46).
Indications for ICP monitoring in patients with TBI have been defined in adults (47) and—perhaps less clearly—children (48). Uncertainty persists regarding the value of ICP monitoring to positively influence outcome in other conditions such as hypoxic-ischemic injuries (49) and hepatic encephalopathy (50).
Evidence for ICP monitoring has been predominantly obtained from studies of adult patients; however, evidence-based pediatric guidelines were published by the Brain Trauma Foundation in 2003 (51) and these were updated in 2012 (48,52) (hereafter referred to as “the pediatric TBI management guidelines”). ICP monitoring is currently recommended for all children with “severe TBI,” which is generally defined as a postresuscitation Glasgow Coma Score (GCS) of ≤ 8. Mild to moderate head injury does not routinely require ICP monitoring, but if the child is unable to be serially neurologically monitored, due to airway management, sedation, and analgesia concerns, then the treating physician may opt to monitor ICP invasively. The presence of an open fontanel and/or open sutures does not preclude the development of raised ICP.
A correlation between raised ICP and poor outcome has been demonstrated in both adult (53,54) and pediatric (55,56) studies of TBI. The purpose of monitoring ICP is to identify, and guide management of, raised ICP (generally defined as ICP ≥ 20 mmHg) (57).
In spite of published recommendations, variability in the implementation of ICP monitoring exists between centers. A prospective study examining ICP monitoring use in the United Kingdom found that overall only 59% of patients presenting to the emergency department with severe TBI (GCS ≤ 8) had ICP monitored, and the use in individual centers varied widely (7% to 100%) (58).
The adult literature is more specific with its recommendations (47), with level II evidence that ICP should be monitored in patients with severe TBI and a GCS between 3 and 8 following resuscitation and an abnormal computed tomographic (CT) scan of the head—defined as demonstrating either hematomas, swelling, contusions, herniation, or compression of the basal cisterns. There is level III evidence that ICP monitoring is indicated in patients with severe TBI and a normal head CT scan if two of the following three clinical features are noted on admission: age over 40 years, unilateral or bilateral posturing, or a systolic blood pressure < 90 mmHg.
Whether monitoring ICP has an impact on functional neurologic outcome remains uncertain. A retrospective study (59) of adult patients with blunt TBI treated in the United States from 1994 to 2001 actually found a 45% decrease in survival in those patients who had ICP monitoring as part of their treatment. This result needs to be interpreted with caution as treatment modalities subsequently shown to have a detrimental effect on outcome, such as hyperventilation and steroids, may have been used in response to intracranial hypertension. Many clinicians would be reluctant currently to enroll patients with severe TBI into a prospective trial which included randomization to a “no ICP monitoring” arm.
■ ICP MONITORING TECHNIQUES
ICP monitoring devices should be tailored to specific patient groups, but there are some generic characteristics that are favorable for all devices. These characteristics are accuracy, reliability, minimal patient morbidity, and cost effectiveness.
ICP may be measured by direct or indirect methods. Direct methods allow monitoring of true ICP, while indirect techniques allow only assessment of ICP trends.
Direct ICP Measurement
There are 3 types of invasive catheters currently available: (1) the external strain gauge, (2) microstrain gauge, and (3) fiberoptic catheter. Each type has advantages and disadvantages. Devices can be inserted in different locations within the injured central nervous system (CNS). The external strain gauge system relies on a fluid filled catheter in direct communication with the patient’s intracranial cavity. The transducer must be calibrated in relation to a fixed point on the patient’s head (usually the tragus) and adjusted each time the level of the head is moved. These devices have been shown to be reliable (60) and can be easily recalibrated, but air bubbles or obstruction to the catheter can cause inaccuracies. The microstrain gauge and fiberoptic technologies require calibration prior to surgical insertion and once in situ cannot be easily recalibrated. Catheters can be placed in a range of different locations within the neurologic system, and the commonly used sites and modalities are outlined below. Minimum standards for any ICP monitoring were established in the late 1980s by the Association for the Advancement of Medical Instrumentation, and include the ability to measure pressure in the range from 0 to 20 mmHg, accuracy to 2 mmHg within the stated range, and a maximum error of 10% in the range 20 to 100 mmHg.
Intraventricular Devices
Lundberg in 1960 (60) published the first method to directly measure ICP using a fluid filled external strain gauge, and this remains the gold standard (61–64). A limitation of this catheter is that it can be difficult to insert when the ventricles are effaced. Ventricular catheters allow CSF to be drained if there is raised ICP, providing a therapeutic option not shared by other monitoring systems (65). Complications include hemorrhage, infection, and malposition. There is a paucity of studies looking at catheter complications in children, and the majority of the data is derived from adult studies. The rate of hemorrhagic complications in adults varies between 1% (62,66) to 5% (67) and varies according to the level of experience of the proceduralist. Around 0.5% of patients may require hematoma evacuation (62,66,67). The incidence of hemorrhagic complications in children has been reported to be much higher at up to 15% (68). The rate of infective complications has been reported to be up to 5% (69), and is affected by the number of times the catheter is accessed and by the number of days the device is in situ. Finally, the rate of malposition of intraventricular catheters in children has been reported to be as high as 9% (69).
Microstrain gauge and fiberoptic catheters have been placed into the intraventricular space but these catheters do not have any therapeutic options. As a result, they have not been widely adopted as the sole monitors for ICP placed within the intraventricular space, unless the ventricles are effaced.
Parenchymal Devices
The intraparenchymal monitoring devices are available as microstrain gauge or fiberoptic catheters. They have been compared against the gold standard and both the micro strain gauge (63,64) and fiberoptic catheters (61) have been reported to be accurate. These catheters are calibrated prior to insertion into the parenchymal tissue and cannot be recalibrated once inserted. The deviation from the calibrated “zero” after a period of monitoring is called “drift” and has been assessed following removal of the catheter. Both microstrain gauge (64,70) and fiberoptic probes (62,71) show some drift over time, but both are considered clinically acceptable (72,73). The drift is an important factor to consider when aggressively treating ICPs in patients who have had these catheters in situ for over a week. Complication rates for the intraparenchymal catheter are relatively low. Hemorrhagic complications in children using fiberoptic catheters have been reported to be between 0% (74,75) and 6.5% (69). The clinical infection rate of these devices is also low at between 0% (69,74) and 2% (76). The microstrain gauge device has a very similar complication profile with very few hemorrhagic complications and infections reported (64,77). These types of catheters have increased in popularity in pediatric intensive care units due to the relatively low complication profile with clinically acceptable drift, but one of the major limitations is the lack of any therapeutic options. Whether it is optimal to monitor the noninjured parenchymal tissue, the penumbra area, or injured tissue with these devices remains uncertain.
Subdural and Epidural Devices
Epidural and subdural monitoring catheters are rarely used in pediatric patients although their risk profile is low. The subdural device requires anchoring the catheter probe to the skull, which makes it difficult to use in neonates and young infants. Cerebral edema increases the risk of probe malfunction due to brain tissue blocking the lumen of the catheter, and therapeutic CSF drainage is not possible with this device. The epidural device in practice has been found to frequently malfunction, become easily displaced, has significant baseline drift, and provides no therapeutic option, and as a result has not been widely adopted.
Indirect ICP Measurement
Indirect techniques for measuring ICP are mainly used as screening tools and have not replaced invasive real-time assessment of ICP. In spite of being less invasive with a lower risk profile, the measurements obtained are merely surrogates for the ICP and the initiation of aggressive ICP treatment strategies without reliable monitoring of the actual ICP is usually considered inadequate. The majority of indirect methods only monitor ICP intermittently and continuous monitoring with early aggressive intervention aimed at minimizing episodes of raised ICP is still considered the best way to prevent long term neurologic morbidity.
Tonometry
The optic nerve sheath is in direct communication with the CNS so an increase in ICP directly impacts on the perioptic nerve space. This mode of measuring ICP has been used in the acute emergency setting as a screening tool to identify those patients who require more aggressive forms of ICP monitoring and treatment. Studies in adults (78,79) and children (80) have demonstrated a significant correlation between an increase in the optic nerve sheath diameter to 5 and 6 mm and elevated ICP. Ultrasonic assessment of intraocular pressure has also been reported to identify elevated ICP (81).
Visual Evoked Response
Visual evoke response (VER) was used in the early 1980s in an attempt to monitor changes in ICP (82). This method relies on an intact optic pathway and assesses a time latency of a specific wave called the N2 wave following exposure to a visual flash. More recent studies in both children (83) and adults (84) have demonstrated a good correlation between VER and raised ICP. Although noninvasive with a low complication profile, this technique has not yet been adopted widely for patients with raised ICP.
Transcranial Doppler
TCD is used to assess the velocity of blood in blood vessels in the basal intracranial arteries. Studies in children (85) and adults (86) have demonstrated good correlation between changes in blood flow and a rise in ICP (87). This type of monitoring has been used as a screening tool to identify patients who might benefit from more invasive ICP monitoring and treatment. Uncertainty remains regarding the accuracy of this tool in predicting changes in ICP (88) and results appear to be highly operator dependent.
Jugular Bulb Oxygen Saturation Monitoring
This mode of indirect monitoring involves the retrograde insertion of an oximetry catheter into the jugular bulb to detect oxygen desaturation in the venous blood returning from the brain. This is based on the assumption that in the absence of any changes to the cerebral metabolic rate, any decrease in venous oxygen saturation will be secondary to a rise in ICP (89,90). Reports suggest that the use of this mode of monitoring, in conjunction with the use of brain tissue oxygenation monitoring (91), can aid in the early detection of low CPP and may prevent further secondary insult (92,93). As retrograde cannulation of the internal jugular vein is required, it has not been widely adopted in pediatric intensive care units, with a reported use in only 4% of children with suspected raised ICP in the United Kingdom in 2006 (58).
Newer Monitoring Techniques
Brain Tissue Oxygenation
Direct brain tissue oxygenation (PbtO2) monitoring involves a specialized monitoring catheter placed into brain parenchyma measuring real-time partial pressure of oxygen. The majority of studies using this device have been performed in adults; however, there is increasing evidence that minimizing low tissue oxygen levels in the injured brain minimizes secondary damage and improves outcomes in patients with TBI (94–96). Results of pediatric studies are less conclusive about outcome improvements but have indicated that using PbtO2 in conjunction with other modes of monitoring may aid in the treatment of children with TBI (97). Where direct brain tissue monitoring is used, it is recommended that PbtO2 should be maintained ≥ 10 mmHg (98).
Near-Infrared Spectroscopy
Near-infrared spectroscopy (NIRS) is a noninvasive monitoring system that measures the percentage of oxygenated hemoglobin (rScO2) in the interrogated tissue by passing a beam of near-infrared light and calculating the difference between oxygenated and deoxygenated blood. This noninvasive technique offers real-time information, and studies have shown an association between low rScO2 and poor outcome following brain injury (99,100). A limitation is that the relationship between raised ICP and rScO2 may be influenced by the type of brain injury (101) and until a clearer understanding of this relationship is established, NIRS cannot be recommended as a standard ICP monitor.
■ ICP WAVEFORMS
In order to understand and interpret a measured ICP value, two aspects of ICP measurement must be considered.
First, the individual waveform has three peaks called P1, P2, and P3 (Figure 3.1), and these provide varying information. These peaks are associated with the arterial and venous waveforms and changes in their morphology may indicate changes in the compliance within the cranial vault. The P1 wave has a fairly sharp peak with relatively constant amplitude; it is the tallest of the 3 waves and an increase in systolic blood pressure will increase the amplitude (102). The P2 and P3 waves end and follow the dicrotic notch. An increase in the amplitude of P2 either indicates an increase in venous volume or a decrease in compliance within the cranial vault (102).
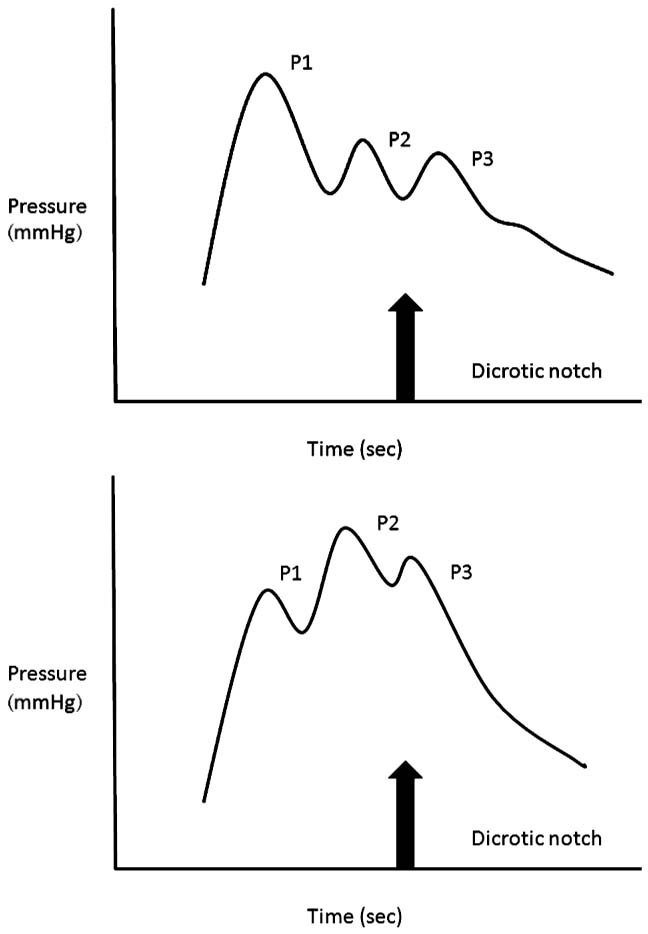
FIGURE 3.1 The intracranial pressure (ICP) waveform.
Second, ICP generates varying patterns over time. In the 1960s Lundberg et al (60,103,104) described three types of wave patterns which they labeled “A”, “B,” and “C” (Figure 3.2). The “C” wave is the result of normal variation in the cardiac cycle and is not considered pathologic. Wave “B” is characterized by a rise in ICP to 30 to 50 mmHg for 5 to 10 minutes before returning to the baseline level. This wave pattern is considered pathologic if there more than three events in a 24 hour period. Wave “A” is a pathologic pattern characterized by a rise in ICP to above 50 mmHg for as long as 20 minutes and failing to return to the pre-event baseline.
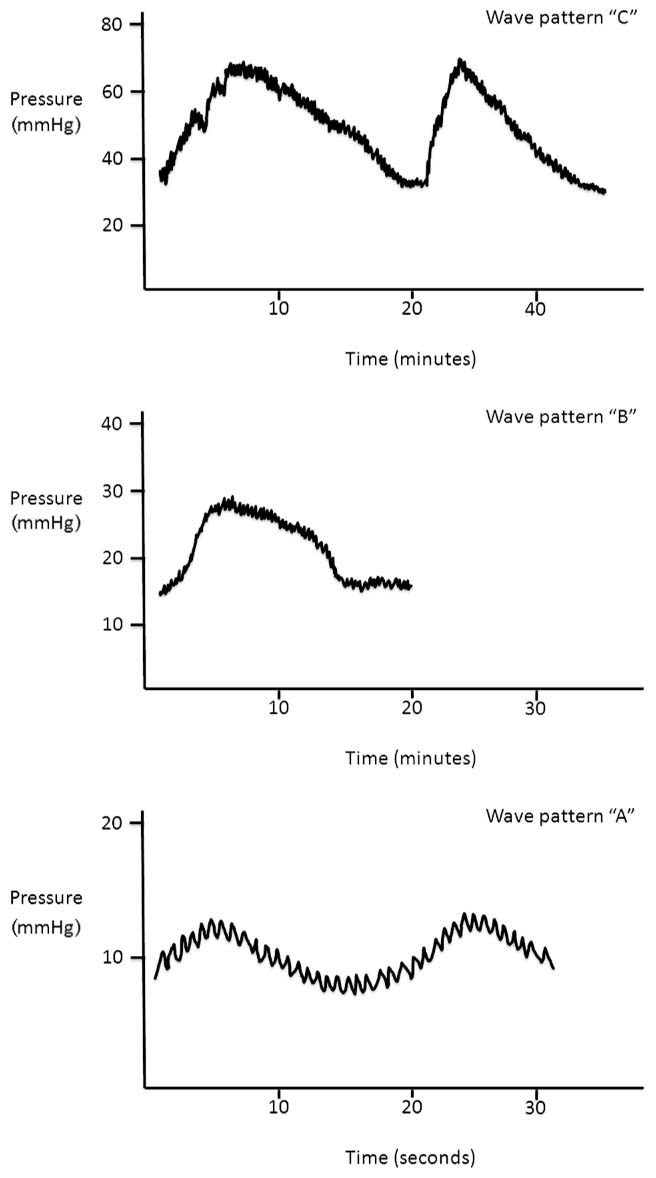
FIGURE 3.2 ICP wave patterns.
■ MANAGEMENT OF RAISED ICP
Raised ICP has been consistently identified as an independent marker of a poor prognosis (18,54,105). As a consequence, ICP has long been used as a surrogate therapeutic target with management strategies largely focused on interventions to either prevent the development of raised ICP, or, when the ICP becomes elevated, to safely lower the ICP to a more “acceptable” (less harmful) range. Conclusive evidence that such strategies improve mortality and neurologic outcome are, however, lacking.
The underlying aim of ICP management is to prevent or minimize secondary ischemic insult to the brain by ensuring that oxygen delivery (DO2) remains in excess of cerebral oxygen consumption (CMRO2) at all times. This is best achieved by focusing on delivering good general intensive care, attending to the “airway, breathing, and circulation” (“ABC”) of resuscitation, and optimizing CBF.
Patient management goals should be to (1) ensure adequate ventilation, oxygenation, and cardiac output, (2) optimize cerebral perfusion by either decreasing ICP or increasing the arterial blood pressure, (3) decrease cerebral metabolic requirements, and (4) ensure that adequate supplies of oxygen and glucose are available in the blood.
It follows from the Monro-Kellie doctrine referred to above that an ICP reduction may be achieved by either decreasing the volume of any of the intracranial contents (brain, CSF, blood, or pathologic space occupying mass), or increasing the size of the cranial compartment. Treatments to lower the ICP therefore might include: reducing the amount of interstitial fluid (edema) in the brain tissues (hyperosmolar therapy, manipulation of the microcirculation [eg, Lund approach]); decreasing the amount of intracerebral blood (hyperventilation, hyperosmolar therapy); decreasing the amount of CSF (drainage via a ventriculostomy); evacuating space-occupying masses/hematomas; and, finally, by increasing the volume of the cranial compartment (decompressive craniectomy).
No single intervention is effective in all patients and given the lack of evidence for many of the available interventions, it is logical that interventions are introduced in a stepwise or “tiered” sequence such as that outlined in Figure 3.3. Tier 1 interventions are generally less invasive with fewer side effects, and are therefore implemented before those Tier 2 interventions that are often more invasive and carry increased risks of causing harm (106).
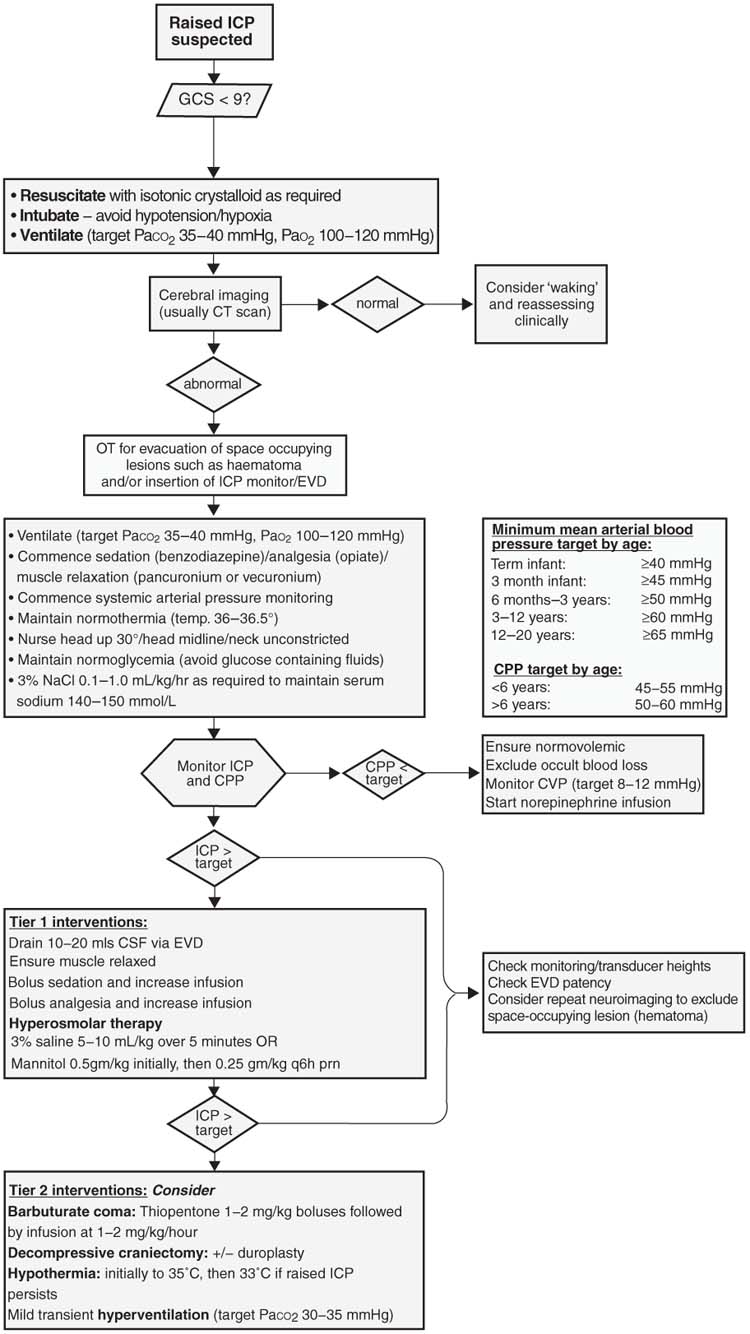
FIGURE 3.3 Suggested algorithm for the management of raised ICP.
■ GENERAL MEDICAL MANAGEMENT
Initial Stabilization and Resuscitation
Patients with acute intracranial hypertension will often present with Cushing triad which is comprised of irregular respiration, hypertension, and bradycardia. Decreased level of consciousness and impaired airway protective reflexes are often present. Patients with TBI as a component of multisystem trauma may have hypotension due to associated hypovolemia or spinal cord injury. Many are also hypoxemic at presentation (107). A systematic ABC-style approach to resuscitation such as that taught in pediatric advanced life support courses requires focus on the rapid assessment and stabilization of the patient’s airway (with cervical spine precautions), breathing, and circulation.
Induction and Intubation
All patients suspected of having raised ICP who are obtunded (GCS 8 or less) should be intubated to establish and maintain a secure airway. Secondary cerebral insults such as hypotension and hypoxia, as well as intracranial hypertension that may be seen during intubations—particularly in the partially conscious patient—must be avoided. Therefore induction and intubation in patients with raised ICP should be performed by the most experienced operator available.
Although the depolarizing muscle relaxant succinylcholine (suxamethonium) may cause a slight ICP elevation, this tends to be mild and transient, and the rapidity of onset which provides optimal intubating conditions generally outweighs any potential drawbacks. A defasciculating dose of a nondepolarizing neuromuscular blocker administered prior to succinylcholine prevents the rise in ICP; however, this is seldom used. An intubating dose of a nondepolarizing agent such as rocuronium (0.6–1.2 mg/kg) may be considered as an alternative to succinylcholine. Intravenous lignocaine (1.5 mg/kg) administered 3 minutes prior to intubation may also prevent the transient rise in ICP associated with intubation and may be considered as an adjunct to the procedure (108–110).
Induction agents such as thiopentone and propofol afford excellent cerebral protection during intubation but may cause transient hypotension, particularly in critically ill hypovolemic patients. Alternative agents such as ketamine and etomidate are less likely to cause hemodynamic instability. Etomidate has been associated with adrenal suppression and is best avoided in the patients who are likely to be unwell for some time. Ketamine has been considered contraindicated in patients with possibly raised ICP due to the widely held belief that it can elevate ICP further; however, this dogma has been recently challenged and appears to have little basis (111–113).
Mechanical Ventilation
Mechanical ventilation of the obtunded patient with raised ICP is essential to avoid hypoxemia, hypocarbia, and hypercarbia. Mechanical ventilation is not a benign intervention, however, and the sedation and muscle relaxation often used to facilitate it are associated with potential complications such as atelectasis and ventilator-associated pneumonia which must be recognized and prevented. Positive intrathoracic pressure can also impede venous return, including that from the head. Although the application of positive end-expiratory pressure (PEEP) may lead to an increase in ICP, this increase appears to be clinically insignificant even at levels as high as 15 cm H2O, with preservation of CPP (114), and we recommend starting with PEEP of 4 to 6 cm H2O. The patient’s minute ventilation should be adjusted to maintain a PaCO2 35 to 40 mmHg (4.7–5.6 kPa).
Chest physiotherapy should be provided as necessary to prevent atelectasis and ventilator-associated pneumonia. Although manual hyperinflation and endotracheal suctioning may increase ICP (115,116), particularly if treatment is begun when the ICP is > 15 mmHg (117,118), percussion can actually lower ICP (117,119). Pretreatment with analgesia, sedation, lignocaine (intravenous or endotracheal) (116), or neuromuscular blockade (118) can attenuate ICP rises and should be considered, and the treatment duration should be as brief as possible.
Volume Resuscitation
Even brief episodes of hypotension can be harmful in the setting of raised ICP and must be strenuously avoided. Patients with TBI may have suffered multitrauma and hypovolemia requiring fluid resuscitation. The choice of prehospital resuscitation fluid in patients with TBI has recently been studied in a randomized controlled trial, and no difference in outcome was identified between the patients resuscitated with hypertonic saline/dextran solution, hypertonic (7.5%) saline, and normal (0.9%) saline (120). These results are consistent with other studies (121), and therefore an isotonic crystalloid solution (normal saline or Hartmann solution) is recommended for prehospital fluid resuscitation.
In terms of in-hospital choice of resuscitation fluids, a recent post hoc subgroup analysis of the multicenter SAFE study (122,123) compared the administration of 4% albumin to crystalloid solution (0.9% saline) for volume resuscitation in adults with TBI. At 2 year follow-up, patients with severe TBI who received albumin had a relative risk of death of 1.88 (95% confidence interval 1.31–2.70, P < .001) compared to those who received normal saline. This finding strongly suggests that crystalloid fluids should be the preferred resuscitation fluid in adults with TBI and in the absence of contradictory evidence this should apply to pediatric practice as well.
Surgery
Once resuscitated and stabilized, any patient with suspected raised ICP should have a cerebral CT scan. A decision must be made regarding whether any significant space occupying lesions or other surgically remedial lesions are present that can be addressed, and whether ICP monitoring is indicated (see above). Although the value of surgical evacuation of significant extraaxial (subdural and extradural) hematomas is well established, a study is currently underway to examine whether evacuation of traumatic intracerebral hematomas is also beneficial (124).
Head Positioning and Posture
In patients with raised ICP, positioning with the head of the bed (trunk and head) elevated is thought to encourage CSF drainage and lower ICP, with a concomitant improvement in CPP. Overall, the empirical evidence supports elevation of the head of the bed by 15° to 30° to decrease ICP and improve CPP (125,126). Elevation of the head of the bed also minimizes the risk of pulmonary aspiration, particularly given that most of these patients will be muscle-relaxed. However, elevating the head places it higher than the heart, and this may decrease the MAP. This potential decrease is proportional to the height of the patient (decreased “pressure” [mmHg] at 30° = 0.5 × distance from the heart
to the head [tragus][cm]/1.36). A recent study in children with TBI found that elevation of the head of the bed led to a lower ICP and MAP in most patients (127). The resultant CPP changes were negligible. Importantly, a paradoxical relationship between head elevation and ICP occurred in some patients, and the authors concluded that the optimal degree of elevation of the head of the bed should be established individually.Venous outflow from the head must be unimpeded by ensuring that the head is in the midline, cervical collars are well fitted, and that any ties used to secure endotracheal tubes are not constricting.
Analgesia and Sedation
Patients with intracranial hypertension may suffer pain from a number of causes, particularly postoperative patients and those with traumatic injuries. Noxious stimuli may cause elevations in the ICP, and anxiety and pain can increase CMRO2 consumption 2- to 3-fold. It is therefore important that all patients receive adequate sedation and analgesia.
A recent systematic review of randomized controlled trials comparing sedation agents for adults with severe TBI failed to find evidence that one drug is more efficacious than any other (128). The issue of analgesia is usually adequately addressed with an opiate infusion such as morphine (10–40 micrograms/kg/hour) or fentanyl (1–2 micrograms/kg/hour).
Adequate sedation is usually achieved with a benzodiazepine, and our usual practice is to commence a midazolam infusion at 1 to 2 micrograms/kg/minute. Intermittent boluses of diazepam or lorazepam may be given as an alternative. Diazepam has been shown to decrease CMRO2 by up to 25% (129). Although propofol is often used in adult patients, long-term use as a sedative in intensive care children is contraindicated due to safety concerns raised by the United States Food and Drug Administration (130) as well as the United Kingdom Committee on Medication Safety (131). Ketamine may be considered in certain situations, and may be helpful if there is hemodynamic instability with other sedatives (113).
Anticonvulsants
Seizures may occur in up to 22% of patients following moderate to severe TBI (132) as well as intracranial hemorrhages. Many of these seizures may be nonconvulsive and only detected by continuous electroencephalographic (EEG) monitoring. In addition, many patients with intracranial hypertension will be muscle-relaxed making seizure activity subtle or absent, and seizures may easily go unrecognized despite close clinical observation.
Benzodiazepines have the added advantage of having some anticonvulsant effects, although patients with either documented seizures or a high risk of seizures should receive prophylactic treatment with phenytoin. Phenobarbitone may be a reasonable alternative anticonvulsant in infants, and levetiracetam may be given intravenously in older children if phenytoin fails to adequately control seizures. A recent study by Vespa et al (133) found that despite therapeutic phenytoin levels, a significant number of adults with severe TBI experienced nonconvulsive seizures which were associated with elevated ICP as well as increased lactate to pyruvate ratios on microdialysis monitoring suggesting possible ischemia. Patients in this study with documented seizures also received levetiracetam; however, they uniformly required induction of burst suppression with midazolam, pentobarbital, or propofol before the seizures were adequately controlled.
The recent pediatric TBI management guidelines recommend that anticonvulsants be considered following severe TBI to reduce the incidence of posttraumatic seizures (134).
Sedation and Analgesia for Interventions
Bolus medications to blunt an anticipated rise in the ICP associated with planned interventions such as endotracheal suctioning and chest physiotherapy may be useful. Potentially useful agents may include narcotics, benzodiazepines, propofol, lignocaine (116), or even ketamine (113). Care must be exercised with bolus opiates as there is a risk these may increase ICP and decrease CPP (128).
For refractory intracranial hypertension, induced anesthesia with a thiopentone infusion will usually decrease ICP as well as decrease the cerebral metabolic rate of oxygen (CMRO2). Such treatment is not without complications, however, and should be reserved for use as a Tier 2 intervention (see below). Monitoring with continuous or intermittent EEG is useful. Following prolonged infusion, plasma thiopentone levels can be monitored. Following prolonged infusions, thiopentone may accumulate in the body’s fat stores and recovery may be delayed.
Neuromuscular Blockade
By producing relaxation of the chest wall and abdominal muscles, neuromuscular blockade decreases intrathoracic pressure, facilitates mechanical ventilation, and prevents patient-ventilator dysynchrony and coughing, all of which may decrease ICP. The use of neuromuscular blockers is not without risk, however, and they should be discontinued as soon as possible.
Glucose and Nutritional Modifications
Following neurologic injury resting energy expenditure has been estimated to be markedly elevated (135). Several authors have addressed the timing of nutrition and a Cochrane Review concluded that early feeding is associated with a trend towards improved survival in adult patients with head injuries (136). The mode of nutrition is also important. Compared to parenteral nutrition, enteral nutrition may reduce complication rates and improve outcome in patients with neurologic injury (137). Children have significantly different metabolic requirements than adults and extrapolating conclusions from adult studies is inadequate. Further studies are needed to determine the optimal needs and timing of nutrition in children with neurologic injuries.
Although glutamine has been found to be beneficial in general trauma patients (138) it has not been specifically studied in either TBI patients or children. Supplements such as zinc (139) and vitamins C and E (140) also require further investigation to determine whether they have a role in improving neurologic outcome in children with neurologic injuries. The pediatric TBI management guidelines found no evidence to support the use of an immunomodulating diet in TBI patients (141).
Finally it remains unclear how hyperglycemia or hypoglycemia affects the injured brain. There are several studies demonstrating hypoglycemic episodes have a negative effect on neurologic outcome (142–144), as well as a substantial body of evidence that indicates that hyperglycemia is an independent risk factor for death in both adults (145–148) and children (148,149) with TBI. Studies in the pediatric population are few and, in view of the real risk of damage to the developing brain of an infant due to hypoglycemia, it is very important to determine if an upper glycemic threshold exists beyond which injury is caused to the brain.
Generally, normal saline (or Ringer lactate/Hartmann solution) should be provided as maintenance fluids in the first instance, the patient’s blood glucose should be monitored frequently to avoid episodes of hypoglycemia and hyperglycemia, and enteral nutrition should be commenced as soon as possible.
■ TREATMENT THRESHOLDS
Patients with a higher measured ICP have been demonstrated to have a worse outcome (death or neurologic morbidity) (18,54,105). Although it is recognized that intracranial hypertension in this setting may represent an epiphenomenon rather than be causally related to the poor outcome, the consistent association between elevated ICP and poor outcome has led to efforts to decrease ICP, and 20 mmHg has been broadly accepted as a threshold for treatment escalation in both adults (150) and children (151). Transient elevations may be tolerated, and many protocols stipulate a period of approximately 5 minutes before advising intervention. It is suspected that a lower ICP threshold should apply to younger children (152); however, the exact levels remain unclear and for the time being, at least, the consensus is that 20 mmHg be considered a reasonable threshold for treatment (151).
The focus on ICP is based on the understanding that an elevated ICP will impede cerebral perfusion. As discussed above, CPP is calculated as the difference between the MAP and mean ICP (CPP = MAP – ICP). The minimal level of “adequate” CPP in the adult population remains uncertain; however, an attempt to balance the risks of using aggressive measures to elevate MAP (such as acute respiratory distress syndrome) against the risks of cerebral hypoperfusion has led to a range of 50 to 70 mmHg being currently recommended (153).
For pediatric patients the ideal target CPP is even less clear, but the pediatric TBI management guidelines recommend that CPP should be maintained greater than 40 mmHg (154,155). It has been demonstrated that the secondary insults leading to adverse outcomes may be “dose responsive,” that is, proportional to both the amount and duration the ICP is above or CPP is below the identified threshold (156).
Target the ICP or the CPP?
In the 1990s two somewhat divergent strategies emerged aimed at minimizing cerebral insults associated with raised ICP following TBI. The CPP-targeted strategy emphasized measures to maintain CPP above a target value of 70 mmHg based on the belief that a low CPP would lead to a decrease in CBF and cerebral hypoperfusion, which in turn results in a “spiral” involving reflex vasodilation, increased cerebral blood volume, raised ICP, and further decreased CPP (157). Therapy to stabilize the CPP included the use of vasopressor agents such as phenylephrine or norepinephrine to increase the systemic blood pressure, euvolemia or mild hypervolemia, and ICP-lowering measures such as CSF drainage and mannitol administration. Rosner et al (157) reported the initial results using this strategy on 158 adult patients following severe TBI. Over the first 10 days of treatment the mean ICP for the study population was 27 mmHg and mean CPP 83 mmHg. Overall mortality in the group treated was 29%, with a favorable outcome (Glasgow Outcome Score [GOS] = 4–5) in 59%. There was no control group in the study; however, when compared to matched patients from the Traumatic Coma Data Bank, both mortality and morbidity outcomes were significantly better in the CPP-targeted patients. The 40% of patients who received vasopressors in this study had a lower GCS and higher ICP at admission compared to those who did not, and the mortality rate was over twice as high (48% vs. 18%). Half the deaths were attributed to Acute Respiratory Distress Syndrome (ARDS), renal failure, sepsis, and pathologies other than neurologic.
The second approach—referred to as “Lund therapy” (158)—targeted a low ICP while accepting a CPP of 50 mmHg in adults (and 40 mmHg in children). This approach aims to decrease cerebral volume by encouraging the reabsorption of water across the transcapillary membrane by preserving the colloid oncotic pressure and decreasing the intracapillary hydrostatic pressure. The latter objective is achieved by decreasing the systemic blood pressure using a combination of a β1-antagonist (metoprolol) and an α2-agonist (clonidine), and also producing moderate constriction of the precapillary resistance vessels using low-dose thiopental and dihydroergotamine. Dihydroergotamine also causes venous vasoconstriction which might also contribute to a decrease in ICP. The treatment protocol also includes furosemide administration as necessary to achieve a neutral or moderately negative fluid balance, and red blood cell transfusions and albumin as necessary to achieve a hemoglobin 125 to 140 g/L and serum albumin 40 g/L. Eker et al (159) reported their center’s experience using Lund therapy [more recently referred to as the “Lund concept” (44)] in 53 adult patients with raised ICP (> 25 mmHg) following TBI. The results were excellent, with mortality rate of 8% and an overall favorable outcome (GOS 4–5) in 79% [compared to a mortality rate of 29% and “favorable outcome” of 59% in the series reported by Rosner et al (157)]. This also represented a significant improvement compared to a group of historical controls treated in the same unit (47% mortality and GOS 4–5 42%). Lund therapy has also been used in children following TBI, with similarly good outcomes (93% survival, and 80% survivors GOS 4–5) (160).
Robertson et al (161) randomized 189 adults with severe TBI to either an ICP-targeted or CBF-targeted management strategy. The CPP target was ≥ 50 mmHg in the ICP-target group, compared to ≥ 70 mmHg in the CBF-target group. Hyperventilation was permitted in the ICP-target group, but not in the CBF-target group. Measured outcomes included jugular venous desaturation (sjvO2 < 50% for > 10 minutes), refractory intracranial hypertension, and functional outcome. After adjustment for potential confounding factors, the patients in the ICP-targeted group had a 2.4 times higher risk of having an episode of jugular venous desaturation than the CBF-targeted patients and the episodes were of longer duration. Despite these differences, there was no significant difference between the groups in terms of functional neurologic outcomes at 3 and 6 months. The incidence of refractory intracranial hypertension was also similar between the groups. Possibly as a result of therapy, the CBF-targeted group suffered a significantly increased incidence of ARDS (15% vs 3.3%, P = .007).
Despite the impressive outcomes reported from single centers, the implementation of Lund therapy has not become widespread, and CPP targeting remains the predominant strategy in the majority of units.
Norepinephrine produces a more reliable and predictable increase in blood pressure compared to dopamine (162) and is the preferred vasopressor.
■ TIER 1 INTERVENTIONS
CSF Drainage
Removal of CSF from within the cranial compartment, usually by way of an external ventricular catheter, is an efficient and rapid means of decreasing ICP.
In patients who have chronic “shunted” hydrocephalus and develop an acute shunt dysfunction with signs of herniation (apnea, anisocoria, bradycardia, and hypertension), rapid “decompression” may be achieved by “tapping the shunt” (or reservoir). Following sterile preparation of the area to be needled, a 25 gauge butterfly needle is introduced into the purpose-designed area of the shunt device or the Rickham reservoir and the CSF pressure is measured using a manometer. If elevated pressure is confirmed, then approximately 10 mL of CSF should be aspirated (163). More CSF can be aspirated if there is no clinical improvement, with intermittent checks of the CSF pressure.
Patients with TBI and potential raised ICP will often have an external ventricular drain (EVD) placed to permit both ICP monitoring as well as CSF drainage. The system must be “closed” to ensure sterility, and CSF may be drained either intermittently in response to elevated ICP, or the EVD may be set at a specific level above the tragus, a site that approximates the foramen of Munro. The conversion factor for mmHg to cm H2O/CSF is approximately 1.36, so placing the drain at a level ~27 cm H2O (CSF) above the tragus, for example, should result in drainage of CSF when the ICP rises above ~20 mmHg.
Complications include hyponatremia from excessive CSF drainage, catheter blockage, infection, and potential “overdrainage.” CSF may not drain in the presence of profound cerebral edema with ventricular effacement. In addition, intermittent ICP assessment may lead to a delay in detection of significant ICP elevations, and a dual monitoring system should be considered if continuous CSF diversion is used (164).
CSF drainage via a lumbar drain may also be considered as an option if intracranial hypertension is refractory to medical management in the presence of a functioning ventricular drain, and appears to be safe when there are open basal cisterns, and focal lesions with mass affect and shift have been excluded (165).
Hyperosmolar Therapy
In the healthy state, the blood-brain barrier is highly impermeable to even very small solutes. This unique property means that increases in plasma osmolality (and not merely colloid oncotic pressure) lead to an increase in the net flux of water into the capillaries in the cerebral circulation. This concept has resulted in a number of osmotically-active substances being used in an attempt to “dehydrate” the brain, thereby decreasing its volume and lowering the ICP (166,167). The “reflection coefficient” referred to earlier in this chapter in regard to Starling forces relates to how readily the molecule diffuses across a membrane and ranges from 0 (very easily) to 1 (none). Substances used therapeutically in the past have included urea (reflection coefficient 0.48), glycerol (reflection coefficient 0.59), mannitol (reflection coefficient 0.90), and sodium chloride (reflection coefficient 1.00) (168).
Mannitol 20% and varying (hypertonic) concentrations of saline are the osmotic agents most widely used currently (Table 3.2). The “dehydrating” properties of these agents were probably overemphasized previously, and more recent studies suggest their use is associated with a range of other potentially beneficial effects including an increase in intravascular volume and decrease in plasma viscosity. They may also reduce endothelial edema, increasing internal blood vessel diameter and thereby decreasing resistance. The accompanying increase in CBF, when autoregulation is intact, has been linked to reflex vasoconstriction (169), another mechanism for the decrease in ICP observed following their administration. Both agents are widely used to treat children with intracranial hypertension (170).
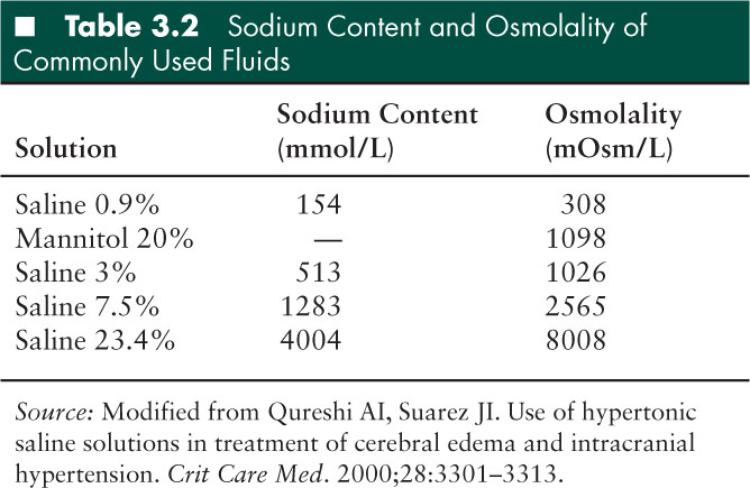
Mannitol
Mannitol has been the mainstay of hyperosmolar therapy for intracranial hypertension for many decades. It is usually available as a 20% solution (20 gm/100 mL), easy to administer, and the recommended initial dose is 0.5 to 1 gm/kg infused over 15 minutes. Further doses of 0.25 to 0.5 gm/kg can be given as long as serum osmolality remains < 320 mOsm/kg, or the osmole gap remains < 10 mOsm/kg. Plasma osmolality can be calculated using the formula (171):
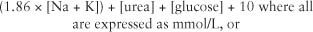

The osmole gap can be calculated by subtracting the calculated osmolarity (mOsm/L) from the measured osmolality (normal range = 0–5) and reflects the quantity of unmeasured osmoles (mOsm/kg). While mannitol is being administered, the osmole gap correlates to some extent with the mannitol concentration, and although the correlation is far from precise, if the osmole gap is low, one can feel reassured that mannitol is being cleared and an additional dose can be safely administered (171).
Potential side effects of mannitol include hypovolemia (due to the induced osmotic diuresis), acute renal failure, hyperkalemia, hypotension, and rebound intracranial hypertension. Historically acute renal failure may have been due to acute tubular necrosis secondary to hypovolemia, and is rarely seen now that patients are generally maintained in a euvolemic state.
Hypertonic Saline
Hypertonic saline was one of the electrolyte solutions used by Weed and McKibben (166,167) in 1919 to demonstrate the potential for hypertonic solutions to decrease the ICP apparently by dehydrating the brain.
Hypertonic saline use has experienced a recent resurgence (172). Initially this was for minimal volume fluid resuscitation of hypervolemic patients, particularly following trauma; however, much research over the past decade has focused on its utility for the treatment of raised ICP. The reflection coefficient for sodium is higher than mannitol, and several studies have found hypertonic saline superior to mannitol (173) leading to suggestions that hypertonic saline should be considered the preferred hyperosmolar agent for the treatment of intracranial hypertension (173–175).
Reported systemic adverse effects include hyperosmolarity, hypernatremia, congestive cardiac failure, hypokalemia, hyperchloremic acidosis, coagulopathy, phlebitis, and renal failure (176). Adverse neurologic effects may include decreased level of consciousness, rebound intracranial hypertension, seizures, central pontine myelinolysis, and subdural and parenchymal hemorrhage (176).
Hypertonic saline is recommended in the recent pediatric TBI management guidelines for the treatment of severe TBI associated with intracranial hypertension (175). Three percent sodium chloride solution (3% NaCl) can be administered as either a slow bolus of 5 to 10 mL/kg, or as an infusion of 3% NaCl solution at a dose range of 0.1 to 1.0 mL/kg/hour. A continuous infusion may be associated with hypernatremia without additional ICP control, and intermittent bolus doses as required may be preferable (177). A recent retrospective study of ICU patients (178) found an association between hypernatremia and ICU mortality. A causal relationship was not proven, and the explanation for the finding may be that hypernatremia is a surrogate marker for disease severity or even reflects a possible increased incidence of diabetes insipidus. Until this becomes clear, it would appear prudent to aim to maintain sodium levels < 165 mEq/L and the serum osmolarity < 360 mOsm/L (179).
Hyperventilation
In the presence of preserved PaCO2 vasoreactivity, hyperventilation results in pH-dependent cerebral vasoconstriction with a decrease in ICP and corresponding increase in CPP. Despite a lack of evidence of improved outcomes, these observed positive effects led to hyperventilation being incorporated into ICP management protocols in the past.
However, more recent studies in patients following TBI involving imaging studies such as xenon-enhanced CT and positron emission tomography used to quantify global and regional CBF and brain tissue perfusion have demonstrated in both adult (180) and pediatric (181) TBI patients that any potential benefits of the decrease in ICP and increase in CPP produced by induced hypocarbia are outweighed by the decrease in CBF and resulting increase in regional brain tissue hypoperfusion/ischemia. The PaCO2 threshold for the onset of significant increase in hypoperfused brain tissue appears to lie between 34 and 38 mmHg (4.5–5 kPa) (182). There was no relationship between CBF and ICP or CPP (181). Global indicators of brain perfusion such as SjVO2 and Arteriojugular Venous Difference of Oxygen (AVDO2) monitoring failed to provide a warning when significant regional hypoperfusion developed, and cannot be relied upon to provide an adequate safeguard when hyperventilation is used (180,182).
Based on these findings, routine (“prophylactic”) hyperventilation should be avoided (32) and the use of hyperventilation should be limited to situations where clinical signs (such as the onset of bradycardia, systemic hypertension, and anisocoria) suggest that brain herniation due to refractory intracranial hypertension is imminent.
■ TIER 2 INTERVENTIONS
Therapies such as barbiturate coma, hypothermia, and decompressive craniectomy have been found to fairly consistently lower ICP, making these interventions attractive to treat intracranial hypertension. However, more recent studies looking at “patient-centric” outcomes such as morbidity and mortality have so far failed to demonstrate a beneficial effect. In spite of this lack of evidence of benefit on outcomes, it is understandable that many clinicians feel compelled to intervene to reduce severe intracranial hypertension that we know is associated with a poor prognosis. It is likely that these interventions may be useful in some instances and harmful in others, and it is hoped that further research may be able to identify the specific patients or circumstances where the likely benefits of these interventions outweigh the potential harms.
Barbiturate Coma
High-dose barbiturates such as thiopentone and pentobarbital decrease cerebral metabolism and oxygen consumption, leading to metabolism-regulated cerebral vasoconstriction resulting in decreased ICP. In addition, barbiturates have some free radical scavenging properties which may provide a degree of protection against ischemic damage. They are therefore attractive therapeutic options for patients with intracranial hypertension. Unfortunately, high-dose barbiturate therapy has numerous side effects which may outweigh these benefits, including hypotension due to systemic vasodilation and myocardial depression and pulmonary complications. Despite the fairly consistent effects on ICP, there has been no evidence that the use of high-dose barbiturates is associated with improved outcome in patients with raised ICP (183).
A laboratory-based animal study (26) found thiopentone (producing burst-suppression on EEG) produced a 16% decrease in mean blood pressure, 23% decrease in cardiac index, 45% increase in heart rate, no change in peripheral vascular resistance, and a 38% decrease in cerebral metabolic rate for oxygen. These changes remained constant over a range of PaCO2 levels between 20 and 60 mmHg. The degree of cerebral vasoconstriction induced by barbiturates was approximately equivalent to that seen with a drop in PaCO2 from 40 to 20mmHg.
A number of reports describe unexpectedly good outcomes following the use of barbiturates. Sidi et al (184) described two patients with severe refractory ICP elevation who appeared to respond to thiopentone. Both patients made near-complete recoveries, and although the authors acknowledged this was anecdotal evidence, they recommended thiopentone be considered in patients with refractory ICP.
Pittman et al (185) reported on the use of pentobarbital for elevated ICP in 27 children with TBI. Fourteen patients (52%) had satisfactory control (ICP < 20 mmHg), 6 patients (22%) died within 48 hours, and 7 patients (26%) had extremely high ICP (ICP > 35 mmHg) and a sustained CPP of less than 50 mmHg despite pentobarbital treatment. Six of the 7 patients had a CPP of less than 30 mmHg for several hours. Hypotension was seen in all patients. All 7 patients survived: 3 with a good outcome, 2 moderately disabled, and 2 with vegetative outcomes, and the authors attributed the survival of these patients to beneficial effects of pentobarbital.
In an unpublished study by Bohn et al (186), patients with severe head injuries were randomized to receive either phenobarbitone or “standard therapy” (which at the time included routine steroids and hyperventilation to PaCO2 < 25 mmHg for episodes of raised ICP). There was no difference in terms of mortality, ICP control, or neurologic outcome between the groups at discharge or 6 month follow up.
Other studies have examined whether barbiturates administered prior to the onset of intracranial hyper-tension might somehow offer a degree of protection from ICP elevation and subsequent neurological injury. Ward et al (187) randomized patients older than 12 years with ICP monitoring following head injuries to receive either pentobarbital “prophylactically” to achieve burst suppression on EEG or no pentobarbital. There was no significant difference in hourly levels of ICP between the groups, incidence of patients with elevated ICP, duration of ICP elevation, or the response of ICP elevation to other treatments. This indicated there was no improvement in outcome when pentobarbital was used prophylactically.
Barbiturates should therefore be considered to treat intracranial hypertension refractory to maximal medical and surgical management (188). Thiopentone appears to be more effective at controlling the ICP than pentobarbital (189). The usual dose is 1 to 2 mg/kg as a slow bolus, observing for a decrease in the ICP (and blood pressure). Once ICP control is achieved, an infusion should be commenced at 1 to 5 mg/kg/hour via a central line—extravasation can cause tissue necrosis. EEG monitoring should be performed regularly aiming for a “burst-suppression” pattern. Blood levels can also be monitored. Systemic hypotension is common, and recalling that hypotension has been associated with poor outcomes, it is imperative that it is anticipated and treated promptly with fluid loading and an inotrope (eg, epinephrine) or vasopressor (eg, norepinephrine) infusion as necessary.
Induced Hypothermia
The potential of induced hypothermia to provide cerebral protection for patients with elevated ICP first appeared in the literature over 50 years ago (190–194), and support for its use in clinical practice has waxed and waned over the years since. Therapeutic hypothermia has been demonstrated to have numerous potentially beneficial neuroprotective effects in animal models of TBI (195,196), and has become the subject of many trials over the past decade following evidence of improved outcomes when used for neonates following hypoxic-ischemic injury (197) and adults following out-of-hospital cardiac arrest (198,199).
Following a number of small, single center studies with positive results, a large multicenter study (200) of adult TBI patients (GCS ≤ 8) failed to demonstrate a significant difference in outcomes between groups of patients randomized to hypothermia (initiated within 6 hours of injury, target 33°C) or normothermia, with similar mortality rates (28% and 27% in the hypothermia and normothermia groups respectively), and similar unfavorable neurologic outcomes (dead, vegetative, or severe disability: 57% in both groups). Recruitment was stopped early after 392 patients were randomized (out of a planned 500) due to futility. The investigators found a higher incidence of hypotension in the hypothermia group on days 3 and 4 during rewarming, with a higher mean CPP on day 1 but lower on days 3 and 4. Patients < 45 years of age who were both hypothermic at presentation and assigned to the hypothermia treatment arm had a better outcome than those who were hypothermic at presentation but subsequently rewarmed following assignment to the normothermic arm. This prompted the investigators and others (201) to speculate that the lack of demonstrated benefit may have been due to the hypothermia being instituted too late, and a further trial which aims to enroll patients with 2.5 hours and institute hypothermia within 4 hours of the injury, with strict avoidance of hypotension, has commenced (202).
A further study (Eurotherm3235Trial) is currently underway studying the effect of the rapid institution of hypothermia in patients following severe TBI for the treatment of intracranial hypertension refractory to other (Tier 1) ICP measures (196).
In a recent randomized controlled trial involving 17 centers in 3 countries over nearly 5 years, Hutchinson et al (203) randomized 225 children with severe TBI (GCS ≤ 8) to either the hypothermia (32.5 +/– 0.5°C) or normothermia (37 +/– 0.5°C) treatment group. The cooling had to be commenced within 8 hours of injury and rewarming commenced at 24 hours at a rate of 0.5°C every 2 hours. Somewhat unexpectedly, the hypothermia group had worse outcomes. The proportion of patients with an unfavorable neurologic outcome (the primary outcome) as assessed by the Pediatric Cerebral Performance Score at 6 months was higher in the hypothermia group (32 of 102, or 31%) compared to the normothermia group (23 of 103, or 22%). Mortality was also higher in the therapeutic hypothermia group (23% vs 14%); however, both results failed to reach statistical significance (P values .14 and .06 respectively). There was no difference between groups in other secondary outcomes including duration of ICP monitoring, mechanical ventilation, ICU length of stay, and hospital length of stay. The hypothermia group had a lower ICP (mean 14.7 vs 17.1 mmHg) and lower heart rate (81 vs 108 beats per minute) during the first 24 hours while cooled, and a lower blood pressure (77.7 vs 83.4 mmHg) and lower CPP (60.8 vs 66.0 mmHg) during the rewarming phase (25–72 hours). Based on these findings, the authors concluded that the use of hypothermia following such a protocol is not warranted, but speculated that either earlier or more prolonged cooling might produce benefit. The potentially detrimental impact of hypotension and lower CPP was reinforced in a post hoc analysis of data from the study which confirmed more episodes of hypotension and low CPP in the hypothermia group compared with the normothermia group, and that hypotension and low CPP were associated with an unfavorable outcome in both groups (204).
The Hypothermia in Traumatic Brain Injury in Children (HiTBIC) Trial (ClinicalTrials.gov no. NCT00282269) randomized children with TBI (GCS ≤ 8 and abnormal cerebral CT scan) to be either cooled (32°C–33°C) for 72 hours and then rewarmed slowly (≤0.5°C every 2 hours) or maintained at normal temperature. The results are yet to be published.
The “CoolKids Trial” (ClinicalTrials.gov no. NCT00222742) aimed to examine the effect on mortality and functional outcome of pediatric patients following severe TBI (GCS ≤ 8) randomized to either cooling to 32° to 33°C commencing within 6 hours and lasting at least 48 hours, or the control arm. The rate of rewarming was 1°C every 12 to 24 hours. Recruitment for the study was halted for “futility” after an interim analysis found that continuing the trial was unlikely to provide an answer to the study hypothesis, and the final results and analysis are awaited.
A recent systematic review of evidence for the use of hypothermia in TBI (205) concluded that there was no evidence that hypothermia is beneficial. The pediatric TBI management guidelines also found no evidence to support a level 1 recommendation but suggested that moderate hypothermia (32°C–33°C) may be considered for refractory intracranial hypertension. Based on the Hutchinson trial, however, if cooling is instituted it should be continued for longer than 24 hours (and probably up to 48–72 hours), and the subsequent rate of rewarming should be slower than 0.5°C per hour, with careful monitoring for—and rapid reversal of—hypotension during this phase (206).
Decompressive Craniectomy
When intracranial hypertension is refractory to medical interventions, a decompressive craniectomy, with or without duroplasty, may be performed to enlarge the cranial compartment, thereby relieving ICP. This approach has been used for over a century, and is attractive based on first principles. Despite many case series and anecdotes reporting successful outcomes in both traumatic (207–213) and nontraumatic (214) causes of raised ICP, evidence for the efficacy of this approach is scant (215).
A range of surgical techniques for decompressive craniectomy are described in the literature, including unilateral or bilateral temporal, frontal, frontotemporoparietal, or posterior fossa craniectomies. While the dura mater is not always opened (216), a further significant decrease in ICP (217) as well as increase in PbtO2 (218) following initial craniectomy have been observed following opening of the dura, and many consider dural opening and duroplasty to be a standard of care in this setting.
While the bone flap is usually removed and stored either in a bone bank at –80°C or in a fashioned subcutaneous abdominal pouch, a recent article reported a lower complication rate following a “hinge” craniectomy (219). In this procedure, the bone flap is secured to the surrounding skull by a Y-shaped plate immediately posterior to the coronal suture, with another 2 plates placed to prevent the bone flap subsequently “subsiding” following resolution of the swelling. In addition to fewer complications, this technique also obviates the need for a second procedure.
Complications of decompressive craniectomy may be seen early or late. The decrease in ICP and exposure of the brain to atmospheric pressure has been associated with hyperemia, herniation, venous engorgement with hemorrhagic contusions, and contralateral extraaxial hematomas. Late complications include infections, hydrocephalus, seizures, bone resorption, and CSF circulation abnormalities including CSF hygromas. In addition, a “syndrome of the trephined” has been described consisting of headache, dizziness, concentration difficulties, memory problems, and mood disturbances (220). A number of patients have also developed late onset reversible monoplegia usually affecting the contralateral upper limb (221).
Up until recently, the only published randomized controlled trial involved 27 pediatric patients with refractory raised ICP randomized to decompressive craniectomy or “routine medical management” which included 24 to 36 hours of induced hypothermia to 33°C (216). The decision to randomize was based on an ICP on day 1 of 20 to 24 mmHg for 30 minutes, 25 to 29 mmHg for 10 minutes, ≥ 30 mmHg for 1 minute, or clinical evidence of herniation. This study demonstrated both better ICP control following decompressive craniectomy as well as improved outcome—as assessed by the GOS (222) and Health State Utility Index at 6 month follow up.
A recently published study (223) known as the DECRA trial evaluated outcomes of 155 adults aged 15 to 59 years with diffuse TBI and refractory intracranial hypertension (defined as > 20 mmHg for > 15 minutes within a 1 hour period) randomized to either decompressive craniectomy or standard care. There was a significant decrease in ICP following craniectomy; however, patient outcomes at six months, assessed by the Extended GOS, were actually significantly worse in the decompressive craniectomy group compared to the patients who received standard care. There was no difference in mortality between groups (18% in decompressive craniectomy group vs 19% in standard care group). It should be noted that more patients in the decompressive craniectomy group had unreactive pupils prior to enrollment, and following adjustment for this factor, the differences in outcome failed to reach statistical significance. Also, the indication for decompressive craniectomy in the study of 15 minutes with ICP > 20 mmHg may not be widely accepted. Although it is important not to delay decompressive craniectomy in the setting of possible cerebral hypoperfusion/ischemia, many units would wait longer until proceeding to decompressive craniectomy. In fact, although the ICP in the “standard care” group remained significantly higher than the decompressive craniectomy group, it was generally less than the 20 mmHg threshold following randomization. These limitations have led some to question the generalizability of the study’s findings (224,225).
The RESCUEicp Trial (International Standard Randomised Controlled Trial Number Register no. 66202560) is an ongoing multicenter international randomized controlled trial with 324 of a projected 400 subjects already recruited (226). The definition of “refractory raised ICP” for the purposes of randomization within this study is an ICP > 25 mmHg for > 1 to 12 hours despite optimal medical management (227). Recruitment is apparently proceeding well (228) and a result is expected in the next few years. Given the DECRA study was confined to patients 15 to 59 years of age, and RESCUEicp study looks at patients 10 to 65 years, it remains to be seen whether the results of these studies will be accepted as applicable to the pediatric population.
For now, it is reasonable to consider decompressive craniectomy for patients with raised ICP and signs of neurologic deterioration, including herniation, or intracranial hypertension refractory to other measures (229).
■ FUTURE DIRECTIONS
Although neurologic injury is the leading cause of morbidity in the pediatric population the impact that has been made on long term neurologic outcome has not been encouraging and therefore clinicians are striving to improve treatment strategies. Below, a number of new therapeutic concepts are outlined that either remain in the experimental realm or have been adopted in centers but have not become standard of care.
Newer Pharmacologic Interventions
Progesterone
Progesterone is a hormone that has been demonstrated to have neurosteroidal and neuroactive properties in the CNS. There has been significant interest in the role progesterone has in modifying cerebral edema in the setting of TBI. Several different animal models have been used to investigate this and consistently have shown that there is a reduction in ICP and improvement of neurologic scores (230–232). There is little data on humans with only 3 randomized controlled studies in adults subjects with TBI treated with progesterone. Results are so far encouraging with improved neurologic outcomes at 1 month (233) and 6 months (234) and decreased mortality. Patient numbers are small, however, and a Cochrane Review (235) concluded that larger randomized controlled trials are needed before progesterone can be recommended as a neuroprotective treatment. Furthermore the safety and efficacy of using progesterone in prepubertal children needs to be established before this therapy can be considered in children.
Aquaporin-Channel Modifying Agents
Aquaporins (AQPs) are a relatively recently described family of water channel proteins that appear to regulate water transport in and out of cells. To date, 13 AQPs (236) have been described in mammals but one—AQP4—is considered to be the principal water channel of the cells in the brain. AQP4 is expressed mainly on astrocyte foot process, in particular the cell surface of the blood-brain and CSF-brain barrier (237).
The role that this water channel has in the formation of cerebral edema has been investigated using several animal models. The pathophysiologic mechanisms leading to cerebral edema can be classified broadly into three processes: cytotoxic, vasogenic, and hydrocephalic. The role of AQP4 in the development of cerebral edema has been studied in AQP4 channel deficient (knockout) mice. The cerebral injury model used in these mice differentiated between a vasogenic process (hemorrhagic stroke) and cytotoxic process (ischemic stroke). While a lack of AQP4 expression was found to be beneficial following a cytotoxic injury, resulting in decreased brain water content, infarct size, and lower ICP with improved survival (238), AQP4 deficiency was detrimental in the vasogenic model and associated with increased brain water content, increased ICP, and worse neurologic outcome (239). In pathologies driven mainly by cytotoxic injury, such as hypoxic-ischemic injury, TBI, meningitis, and metabolic derangements, administration of a specific therapeutic agent to inhibit the action of AQP4 might therefore represent a novel way to limit and possibly reduce cerebral edema.
Arginine-vasopressin has been identified as a possible AQP4 modulator. At this stage, studies of an arginine-vasopressin (V1a) receptor inhibitor in both TBI (240) and hemorrhagic brain injury (241) have been limited to animal models; however, the results of these studies show a reduction in cerebral water content and improved neurologic outcomes. Although these early results are encouraging, further studies are needed before AQP-targeted interventions can be trialed in human subjects.
Erythropoietin
Human recombinant erythropoietin has been shown to have neuroprotective properties but the dose required to exert this effect overstimulates the bone marrow (242). To overcome these side effects carbamylated erythropoietin was used in animal studies with a reduction of cerebral edema and improved functional outcomes reported in both ischemic brain injury (243) and TBI (244) models. There has only been one human study in which 16 patients with TBI were randomized to receive erythropoietin or a control. The results of this study showed no difference in outcome (245), but the study was significantly underpowered and much larger multicenterd randomized controlled trials need to be performed to establish the efficacy of this therapy for cerebral edema.
■ CONCLUSIONS
Many questions regarding intracranial hypertension and its management remain unanswered, and it is important that we acknowledge these areas of uncertainty. A number of robust studies have identified various factors and practices that are associated with poor outcomes, and these must be carefully avoided. These include: hypotension (particularly early), early resuscitation with albumin, hyperthermia, hypocarbia, hyperglycemia and hypoglycemia, hypoxemia, raised ICP, and decreased CPP. Whether an ICP-targeted approach to management is superior to one directed at CPP remains uncertain. The role of so-called Tier 2 treatments such as barbiturate coma, decompressive craniectomy, and hypothermia also remains unclear, for although these interventions have been demonstrated to decrease ICP, recent studies looking at longer-term patient-centric outcomes suggest that they may, in fact, cause harm. These findings remind us that we must not focus merely on surrogate markers or endpoints (246,247), and it is too simplistic to assume that an intervention that produces a decrease in ICP or an increase in CPP will necessarily benefit our patient. As with all clinical interventions, we are obliged to “first, do no harm,” and must therefore ensure that we administer those therapies—and only those therapies—that we believe, based on all the available evidence, are likely to provide more benefits than harms to our patients.
■ REFERENCES
1. . The Monro-Kellie hypothesis: applications in CSF volume depletion. Neurology. 2001;56(12):1746–1748.
2. , . Intracranial elastance versus intracranial compliance: terminology should agree with that of other disciplines. Anesthesiology. 1992;77(2):403.
3. et al. Acute management of acquired brain injury: an evidence-based review of pharmacological interventions. Brain Inj. 2010;24(5):706–721.
4. , . The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J Clin Invest. 1948;27(4):476–483.
5. et al. Transcranial Doppler measurement of middle cerebral artery blood flow velocity: a validation study. Stroke. 1986;17(5):913–915.
6. et al. Reproducibility of transcranial Doppler sonography: a validation study. Ultrasound Med Biol. 1992;18(2):173–177.
7. et al. The development of cerebral perfusion in healthy preterm and term neonates. Neuropediatrics. 2003;34(6):281–286.
8. , . A longitudinal study of cerebral blood flow over the first 30 months. Pediatr Res. 2009;66(5):560–564.
9. , . Age dependence of total cerebral blood flow volume from childhood to adulthood. J Cereb Blood Flow Metab. 1996;16(5):827–833.
10. , , . Cerebral blood flow and vascular physiology. Anesthesiol Clin North Am. 2002;20(2):247–264, v.
11. . Positron emission tomography: principles and applications in pediatrics. Mead Johnson Symp Perinat Dev Med. 1987(25):15–18.
12. . Morphometric study of human cerebral cortex development. Neuropsychologia. 1990;28(6):517–527.
13. et al. The cerebral circulation during sleep: regulation mechanisms and functional implications. Sleep Med Rev. 2002;6(6):443–455.
14. , . Cerebral blood flow and metabolism: effects of anesthetic drugs and techniques. Anesthesiology. 1972;36(4):378–400.
15. et al. Variation in cerebral blood flow velocity with cerebral perfusion pressure > 40 mmHg in 42 children with severe traumatic brain injury. Crit Care Med. 2009;37(11):2973–2978.
16. et al. Assessment of cerebrovascular autoregulation in head-injured patients: a validation study. Stroke. 2003;34(10):2404–2409.
17. et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34(2):216–222.
18. et al. Age-related differences in intracranial pressure and cerebral perfusion pressure in the first 6 hours of monitoring after children’s head injury: association with outcome. Childs Nerv Syst. 2005;21(3):195–199.
19. et al. Changes in extracellular calcium activity in cerebral ischaemia. J Cereb Blood Flow Metab. 1981;1(2):203–209.
20. et al. Assessment of the lower limit for cerebral perfusion pressure in severe head injuries by bedside monitoring of regional energy metabolism. Anesthesiology. 2003;98(4):809–814.
21. , . The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest. 1948;27(4):484–492.
22. , The cerebral vascular response to reduction in arterial carbon dioxide tension. J Clin Invest. 1961;40:1297–1303.
23. , . Step hypocapnia to separate arterial from tissue PCO2 in the regulation of cerebral blood flow. Circ Res. 1967;20(2):272–278.
24. , . Effect of alterations in the arterial carbon dioxide tension on the blood flow through the cerebral cortex at normal and low arterial blood pressures. J Neurol Neurosurg Psychiatry. 1965;28(5):449–452.
25. et al. Enhancement of cerebrovascular effect of CO2 by hypoxia. Stroke. 1980;11(3):286–289.
26. et al. Influence of changes in arterial pCO2 on cerebral blood flow and metabolism during high-dose barbiturate therapy in dogs. J Neurosurg. 1981;54(5):615–619.
27. , , . The effects of PaCO2 reduction on regional cerebral blood flow in the acute phase of brain injury. Acta Anaesthesiol Scand. 1977;21(5):359–367.
28. et al. Measurement of vascular reactivity in head injured patients. Acta Neurochir Suppl (Wien). 1993;59:18–21.
29. et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75(5):731–739.
30. et al. Cerebrovascular response in infants and young children following severe traumatic brain injury: a preliminary report. Pediatr Neurosurg. 1997;26(4):200–207.
31. et al. Guidelines for the management of severe traumatic brain injury: hyperventilation. J Neurotrauma. 2007;24(suppl 1):S87–S90.
32. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): hyperventilation. Pediatr Crit Care Med. 2012;13:S58–S60.
33. , , . Interaction of myogenic mechanisms and hypoxic dilation in rat middle cerebral arteries. Am J Physiol Heart Circ Physiol. 2002;283(6):H2276–H2281.
34. et al. Effects of high atmospheric pressure and oxygen on middle cerebral blood flow velocity in humans measured by transcranial Doppler. Stroke. 1998;29(1):94–97.
35. et al. Independent cerebral vasoconstrictive effects of hyperoxia and accompanying arterial hypocapnia at 1 ATA. J Appl Physiol. 2003;95(6):2453–2461.
36. . Description of the myogenic hypothesis. Circ Res. 1964;15(suppl):279–287.
37. , , . Neuronal messengers in the human cerebral circulation. Peptides. 2001;22(6):995–1007.
38. et al. Changes in cerebral blood flow estimated after stellate ganglion block by single photon emission computed tomography. J Auton Nerv Syst. 1995;50(3):339–346.
39. et al. Cerebral blood flow velocities and trigeminal ganglion stimulation. A transcranial Doppler study. Stereotact Funct Neurosurg. 1996;66(4):184–192.
40. et al. Evaluation of cerebrovascular CO2-reactivity and autoregulation in patients with post-traumatic diffuse brain swelling (diffuse injury III). Acta Neurochir Suppl. 1998;71:233–236.
41. et al. Cerebral autoregulation in pediatric traumatic brain injury. Pediatr Crit Care Med. 2004;5(3):257–263.
42. et al. Neurointensive care; impaired cerebral autoregulation in infants and young children early after inflicted traumatic brain injury: a preliminary report. J Neurotrauma. 2007;24(1):87–96.
43. . Quantifying cerebral autoregulation in health and disease. Crit Care Resuscitation. 2004;6(1):59–67.
44. . The “Lund concept” for the treatment of severe head trauma–physiological principles and clinical application. Intensive Care Med. 2006;32(10):1475–1484.
45. et al. Papilledema after acute head injury. Neurosurgery. 1985;16(3):357–363.
46. et al. The diagnosis of head injury requires a classification based on computed axial tomography. J Neurotrauma. 1992;9(suppl 1):S287–S292.
47. et al. Guidelines for the management of severe traumatic brain injury: Indications for intracranial pressure monitoring. J Neurotrauma. 2007;24(suppl 1):S37–S44.
48. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): indications for intracranial pressure monitoring. Pediatr Crit Care Med. 2012;13:S11–S17.
49. et al. Intracranial pressure monitoring following hypoxic-ischaemic cerebral insults. Childs Nerv Syst. 1989;5:280.
50. , . Intracranial pressure monitoring in acute liver failure. A procedure with clear indications. Hepatology. 2006;44(2):504–506.
51. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children and adolescents. Pediatr Crit Care Med. 2003;4(3):S1–S74.
52. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition). Pediatr Crit Care Med. 2012;13(1)(suppl):S1–S82.
53. et al. Improved confidence of outcome prediction in severe head injury. A comparative analysis of the clinical examination, multimodality evoked potentials, CT scanning, and intracranial pressure. J Neurosurg. 1981;54(6):751–762.
54. et al. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. Special Supplements. 1991;75(1s):S59–S66.
55. et al. Treatment and outcome of the severely head injured child. Intensive Care Med. 1983;9(1):13–16.
56. et al. Outcome in children with severe head injuries. Childs Nerv Sys. 1985;1(2):109–114.
57. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents: threshold for treatment of intracranial hypertension. Pediatr Crit Care Med. 2003;4(3 suppl):S25–S27.
58. et al. Intracranial pressure complicating severe traumatic brain injury in children: monitoring and management. Intensive Care Med. 2006;32(10):1606–1612.
59. et al. Intracranial pressure monitoring in brain-injured patients is associated with worsening of survival. J Trauma. 2008;64(2):335–340.
60. . Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Scand Suppl. 1960;36(149):1–193.
61. et al. The fiberoptic intraparenchymal cerebral pressure monitor in 244 patients. Surg Neurol. 1996;45(3):278–282.
62. et al. The Camino intracranial pressure device in clinical practice: reliability, handling characteristics and complications. Acta Neurochir. 1998;140(11):1113–1119; discussion 1119–1120.
63. et al. A clinical evaluation of the Codman MicroSensor for intracranial pressure monitoring. Br J Neurosurg. Jun1998;12(3):223–227.
64. , . Clinical experience with the intraparenchymal intracranial pressure monitoring Codman MicroSensor system. Neurosurgery. 2005;56(4):693–698; discussion 693–698.
65. et al. The scientific history of hydrocephalus and its treatment. Neurosurg Rev. 1999;22(2–3):67–93; discussion 94–65.
66. et al. Cerebral monitoring devices: analysis of complications. Acta Neurochir Suppl. 1998;71:47–49.
67. et al. Safety and accuracy of bedside external ventricular drain placement. Neurosurgery. 2008;63(1)(suppl 1):ONS162–166; discussion ONS166–167.
68. et al. Initial head computed tomographic scan characteristics have a linear relationship with initial intracranial pressure after trauma. J Trauma. 2004;56(5):967–972; discussion 972–963.
69. et al. Complications of intracranial pressure monitoring in children with head trauma. J Neurosurg. 2004;101(1 suppl):53–58.
70. et al. Assessment of zero drift in the Codman intracranial pressure monitor: a study from 2 neurointensive care units. Neurosurgery. J2009;64(1):94–98; discussion 98–99.
71. et al. Fiberoptic intraparenchymal brain pressure monitoring with the Camino V420 monitor: reflections on our experience in 163 severely head-injured patients. J Neurotrauma. Apr 2002;19(4):439–448.
72. . Intracranial pressure: current status in monitoring and management. Semin Pediatr Neurol. 1997;4(3):146–155.
73. et al. The Camino intracranial pressure device in clinical practice. Assessment in a 1000 cases. Acta Neurochir. 2006;148(4):435–441.
74. , , . Risk factors of intracranial pressure monitoring in children with fiberoptic devices: a critical review. Surg Neurol. 1997;47(1):16–22.
75. et al. Hemorrhagic complications of intracranial pressure monitors in children. Pediatr Neurosurg. 2003;39(1):27–31.
76. et al. Results and complications of intracranial pressure monitoring in 303 children. Pediatr Neurosurg. 1995;23(2):64–67.
77. et al. A clinical study of parenchymal and subdural miniature strain-gauge transducers for monitoring intracranial pressure. Neurosurgery. 1996;39(5):927–931; discussion 931–922.
78. et al. Non-invasive assessment of intracranial pressure using ocular sonography in neurocritical care patients. Intensive Care Med. 2008;34(11):2062–2067.
79. , , . Ultrasound measurement of optic nerve sheath diameter in patients with a clinical suspicion of raised intracranial pressure. Emerg Med J. 2011;28(8):679–681.
80. et al. Measurement of optic nerve sheath diameter by ultrasound: a means of detecting acute raised intracranial pressure in hydrocephalus. Br J Ophthalmol. 2002;86(10):1109–1113.
81. et al. Correlation of intraocular pressure with intracranial pressure in children with severe head injuries. Pediatr Crit Care Med. 2010;11(5):593–598.
82. et al. Further studies with a noninvasive method of intracranial pressure estimation. Neurosurgery. 1984;14(4):456–461.
83. . Longitudinal stability of visual evoked potentials in children and adolescents with hydrocephalus. Dev Med Child Neurol. 2001;43(2):113–117.
84. , , . Clinical experience with the noninvasive ICP monitoring system. Acta Neurochir Suppl. 2005;95:351–355.
85. et al. Transcranial Doppler can predict intracranial hypertension in children with severe traumatic brain injuries. Childs Nerv Syst. 2011;27(6):979–984.
86. et al. Noninvasive detection of elevated intracranial pressure using a portable ultrasound system. Am J Emerg Med. 2012;30(6):936–941.
87. , , . Transcranial Doppler ultrasonography in raised intracranial pressure and in intracranial circulatory arrest. J Neurosurg. 1988;68(5):745–751.
88. et al. Transcranial Doppler pulsatility index: not an accurate method to assess intracranial pressure. Neurosurgery. 2010;66(6):1050–1057.
89. , , . Detection of cerebral venous desaturation by continuous jugular bulb oximetry following acute neurotrauma. Anaesth Intensive Care. 1995;23(3):307–314.
90. , . Monitoring the injured brain: ICP and CBF. Br J Anaesth. 2006;97(1):26–38.
91. . Multimodal monitoring: head injury management using SjvO2 and LICOX. J Neurosci Nurs. 2004;36(6):332–339.
92. et al. Early SjvO2 monitoring in patients with severe brain trauma. Intensive Care Med. 1999;25(5):445–451.
93. et al. Incidence of intracranial hypertension related to jugular bulb oxygen saturation disturbances in severe traumatic brain injury patients. Acta Neurochir Suppl. 2002;81:285–287.
94. et al. Brain tissue oxygen guided treatment supplementing ICP/CPP therapy after traumatic brain injury. J Neurol Neurosurg Psychiatry. 2003;74(6):760–764.
95. et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J Neurosurg. 2005;103(5):805–811.
96. , , . Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg. 2009;111(4):672–682.
97. et al. Brain tissue oxygen tension monitoring in pediatric severe traumatic brain injury. Part 2: Relationship with clinical, physiological, and treatment factors. Childs Nerv Syst. 2009;25(10):1335–1343.
98. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): advanced neuromonitoring. Pediatr Crit Care Med. 2012;13:S30–S32.
99. et al. Cerebral hypoxia in severely brain-injured patients is associated with admission Glasgow Coma Scale score, computed tomographic severity, cerebral perfusion pressure, and survival. J Trauma. 2004;56(3):482–489; discussion 489–491.
100. , , . Noninvasive cerebral oximetry: is there light at the end of the tunnel? Curr Opin Anaesthesiol. 2010;23(5):576–581.
101. et al. Diagnosis influences response of cerebral near infrared spectroscopy to intracranial hypertension in children. Pediatr Crit Care Med. 2010;11(4):514–522.
102. et al. Intracranial pressure waveform analysis: clinical and research implications. J Neurosci Nurs. 2000;32(5):271–277.
103. , . Leakage as a source of error in measurement of the cerebrospinal fluid pressure by lumbar puncture. Acta Neurol Scand Suppl. 1965;13(pt 1):115–121.
104. , , . Continuous recording of the ventricular-fluid pressure in patients with severe acute traumatic brain injury. A preliminary report. J Neurosurg. 1965;22(6):581–590.
105. , , . ICP and CPP: excellent predictors of long term outcome in severely brain injured children. Childs Nerv Syst. 2008;24(2):245–251.
106. . Management of cerebral perfusion pressure after traumatic brain injury. Anesthesiology. 2001;95(6):1513–1517.
107. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents: prehospital airway management. Pediatr Crit Care Med. 2003;4(3 suppl):S9–S11.
108. , , . Intravenous lidocaine: optimal time of injection before tracheal intubation. Anesth Analg. 1987;66(10):1036–1038.
109. et al. Intravenous lignocaine pretreatment to prevent intraocular pressure rise following suxamethonium and tracheal intubation. Br J Ophthalmol. 1986;70(8):596–598.
110. , . In patients with head injury undergoing rapid sequence intubation, does pretreatment with intravenous lignocaine/lidocaine lead to an improved neurological outcome? A review of the literature. Emerg Med J. 2001;18(6):453.
111. , , . Ketamine for rapid sequence induction in patients with head injury in the emergency department. Emerg Med Australas. 2006;18(1):37–44.
112. , , . Myth: ketamine should not be used as an induction agent for intubation in patients with head injury. Can J Emerg Med. 2010;12(2):154–157.
113. et al. Effectiveness of ketamine in decreasing intracranial pressure in children with intracranial hypertension. J Neurosurg: Pediatr. 2009;4(1):40–46.
114. et al. Effects of varying levels of positive end-expiratory pressure on intracranial pressure and cerebral perfusion pressure. Crit Care Med. 1997;25(6):1059–1062.
115. , , . The effect of nursing interventions on the intracranial pressure in paediatric traumatic brain injury. Nurs Crit Care. 2011;16(2):77–84.
116. , . Intravenously administered lidocaine prevents intracranial hypertension: during endotracheal suctioning. Anesthesiology. 1980;52(6):516–518.
117. , . The effect of respiratory physiotherapy on intracranial pressure, mean arterial pressure, cerebral perfusion pressure and end tidal carbon dioxide in ventilated neurosurgical patients. Physiother Theory Pract. 1993;9(1):3–11.
118. , . The effect of respiratory therapy on intracranial pressure in ventilated neurosurgical patients. Aust J Physiother. 1986;32:107–111.
119. et al. Changes in intracranial pressure associated with chest physiotherapy. Neurocrit Care. 2007;6(2):100–103.
120. et al. Out-of-hospital hypertonic resuscitation following severe traumatic brain injury: a randomized controlled trial. JAMA. 2010;304(13):1455–1464.
121. et al. Prehospital fluid management in traumatic brain injury. Emerg Med Australas. 2011;23(6):665–676.
122. . Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;357:874–884.
123. et al. Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;357(9):874–884.
124. . Surgical Trial in Traumatic intraCerebral Haemorrhage STITCH (trauma). http://research.ncl.ac.uk/trauma.stitch/ Accessed June 12, 2011.
125. et al. Acute management of acquired brain injury: an evidence-based review of non-pharmacological interventions. Brain Inj. 2010;24(5):694–705.
126. et al. Effect of head elevation on intracranial pressure, cerebral perfusion pressure, and cerebral blood flow in head-injured patients. J Neurosurg. 1992;76(2):207–211.
127. et al. Intracranial pressure and cerebral perfusion pressure responses to head elevation changes in pediatric traumatic brain injury. Pediatr Crit Care Med. 2012;13(1):e39–e47.
128. et al. Sedation for critically ill adults with severe traumatic brain injury: a systematic review of randomized controlled trials. Crit Care Med. 2011;39(12):2743.
129. , . Effects on diazepam on cerebral blood flow and oxygen uptake after head injury. Anesthesiology. 1975;43(1):117–122.
130. FDA. FDA advisory against using propofol for prolonged sedation in the PICU. Food and Drug Administration Web site. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm172351.htm/Accessed December 6, 2011.
131. MCA/CSM. Propofol (Diprivan) infusion: sedation in children aged 16 years or younger contraindicated. Curr Probl Pharmacovigilance. 2001;27:10.
132. et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg. 1999;91(5):750–760.
133. et al. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit Care Med. 2007;35(12):2830.
134. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): antiseizure prophylaxis. Pediatric Crit Care Med. 2012;13:S72–S75.
135. . Guidelines for the management of severe traumatic brain injury: nutrition. J Neurotrauma. 2000;17(6–7):539–547.
136. et al. Nutritional support for head-injured patients. Cochrane Database Syst Rev. 2006(4):CD001530.
137. et al. Percutaneous endoscopic gastrostomy in the neurosurgical intensive care unit: complications and outcome. J Parenter Enteral Nutr. 2007;31(6):517–520.
138. et al. Guidelines for the provision and assessment of nutrition support therapy in the adult critically ill patient. J Parenter Enteral Nutr. 2009;33(3):277–316.
139. et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. Jan 1996;13(1):25–34.
140. , . Metabolic responses and nutritional therapy in patients with severe head injuries. J Head Trauma Rehabil. 1998;13(1):11–27.
141. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): glucose and nutrition. Pediatr Crit Care Med. 2012;13:S68–S71.
142. et al. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: a microdialysis study. J Cereb Blood Flow Metab. 2003;23(7):865–877.
143. et al. Lowering of glucose in critical care: a randomized pilot trial. J Crit Care. 2007;22(2):112–118; discussion 118–119.
144. et al. Differential temporal profile of lowered blood glucose levels (3.5 to 6.5 mmol/l versus 5 to 8 mmol/l) in patients with severe traumatic brain injury. Crit Care. 2008;12(4):R98.
145. et al. Glucose control and mortality in patients with severe traumatic brain injury. Neurocrit Care. 2009;11(3):311–316.
146. et al. Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocrit Care. 2009;11(2):151–157.
147. et al. Persistent hyperglycemia in severe traumatic brain injury: an independent predictor of outcome. Am Surg. 2009;75(1):25–29.
148. et al. Relationship between hyperglycemia and outcome in children with severe traumatic brain injury. Pediatr Crit Care Med. 2012;13(1):85–91.
149. et al. Hyperglycemia and outcomes from pediatric traumatic brain injury. J Trauma. 2003;55(6):1035–1038.
150. et al. Guidelines for the management of severe traumatic brain injury: Intracranial pressure thresholds. J Neurotrauma. 2007;24(suppl 1):S55–S58.
151. et al. Guidelines for the Acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): threshold for treatment of intracranial hypertension. Pediatr Crit Care Med. 2012;13:S18–S23.
152. et al. Age-related differences in intracranial pressure and cerebral perfusion pressure in the first 6 hours of monitoring after children’s head injury: association with outcome. Childs Nerv Syst. 2005;21(3):195–199.
153. et al. Guidelines for the management of severe traumatic brain injury: cerebral perfusion thresholds. J Neurotrauma. 2007;24(suppl 1):S59–S64.
154. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents: cerebral perfusion pressure. Pediatr Crit Care Med. 2003;4(3 suppl):S31–S33.
155. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): cerebral perfusion pressure thresholds. Pediatr Crit Care Med. 2012;13:S24–S29.
156. et al. Critical thresholds of intracranial pressure and cerebral perfusion pressure related to age in paediatric head injury. J Neurol Neurosurg Psychiatry. 2006;77(2):234.
157. , , . Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg. 1995;83(6):949–962.
158. et al. Improved outcome after severe head injury with a new therapy based on principles for brain volume regulation and preserved microcirculation. Crit Care Med. 1998;26(11):1881–1886.
159. et al. Improved outcome after severe head injury with a new therapy based on principles for brain volume regulation and preserved microcirculation. Crit Care Med. 1998;26(11):1881–1886.
160. et al. Severe traumatic brain injury in pediatric patients: treatment and outcome using an intracranial pressure targeted therapy—the Lund concept. Intensive Care Med. 2005;31(6):832–839.
161. et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999;27(10):2086–2095.
162. et al. Direct comparison of cerebrovascular effects of norepinephrine and dopamine in head-injured patients. Crit Care Med. 2004;32(4):1049–1054.
163. . Handbook of Neurosurgery. 6th ed. Thieme Medical Publishers; New York, USA. 2006.
164. et al. Intracranial pressure-monitoring systems in children with traumatic brain injury: combining therapeutic and diagnostic tools. Pediatr Crit Care Med. 2011;12(5):560.
165. et al. Controlled lumbar drainage in pediatric head injury. J Neurosurg. 1995;83(3):453–460.
166. , . Experimental alteration of brain bulk. Am J Physiol. 1919;48(4):531–564.
167. , . Pressure changes in the cerebro-spinal fluid following intravenous injection of solutions of various concentrations. Am J Physiol. 1919;48(4):512–530.
168. , . Use of hypertonic saline solutions in treatment of cerebral edema and intracranial hypertension. Crit Care Med. 2000;28:3301–3313.
169. , , . Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984;61(4):700–706.
170. et al. Osmolar therapy in pediatric traumatic brain injury. Crit Care Med. 2012;40(1):208–215.
171. et al. Osmole gap in neurologic-neurosurgical intensive care unit: its normal value, calculation, and relationship with mannitol serum concentrations. Crit Care Med. 2004;32(4):986–991.
172. . Hypertonic saline in critical care: a review of the literature and guidelines for use in hypotensive states and raised intracranial pressure. Anaesthesia. 2009;64(9):990–1003.
173. et al. Hypertonic saline versus mannitol for the treatment of elevated intracranial pressure: a meta-analysis of randomized clinical trials. Crit Care Med. 2011;39(3):554–559.
174. . Hypertonic saline, not mannitol, should be considered gold-standard medical therapy for intracranial hypertension. Crit Care. 2012;16:113.
175. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): hyperosmolar therapy. Pediatr Crit Care Med. 2012;13:S36–S41.
176. et al. Continuous hypertonic saline therapy and the occurrence of complications in neurocritically ill patients. Crit Care Med. 2009;37(4):1433–1441.
177. , . Hypertonic saline in neurocritical care: Is continuous infusion appropriate? Crit Care Med. 2009;37(4):1521–1523.
178. et al. The relation between the incidence of hypernatremia and mortality in patients with severe traumatic brain injury. Crit Care. 2009;13(4):R110.
179. . Sodium and brain injury: do we know what we are doing? Crit Care. 2009;13(5):184.
180. et al. Hyperventilation following head injury: effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med. 2007;35(2):568.
181. et al. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med. 1997;25(8):1402.
182. et al. Effect of hyperventilation on cerebral blood flow in traumatic head injury: Clinical relevance and monitoring correlates. Crit Care Med. 2002;30(9):1950.
183. , . Barbiturates for acute traumatic brain injury. Cochrane Database Syst Rev. 1999;3.
184. et al. Long-term barbiturate infusion to reduce intracranial pressure. Crit Care Med. 1983;11(6):478–481.
185. , , . Efficacy of barbiturates in the treatment of resistant intracranial hypertension in severely head-injured children. Pediatr Neurosci. 1989;15(1):13–17.
186. et al. High-dose barbiturate therapy in the management of severe pediatric head trauma: a randomised controlled trial. Crit Care Med. 1989;17:S118.
187. et al. Failure of prophylactic barbiturate coma in the treatment of severe head injury. J Neurosurg. 1985;62(3):383–388.
188. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): barbiturates. Pediatr Crit Care Med. 2012;13:S49–S52.
189. et al. Pentobarbital versus thiopental in the treatment of refractory intracranial hypertension in patients with traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12(4):R112.
190. . Therapeutic hypothermia in cases of head injury. J Neurosurg. 1959;16(4):407.
191. , . Hypothermia in the treatment of craniocerebral traumatism. J Neurosurg. 1958;15(2):162.
192. . The use of hypothermia in severe head injuries in childhood. Arch Sur. 1959;79(3):362.
193. , . Hypothermia in the treatment of critical head injury. CMAJ. 1962;87(17):887.
194. . The use of hypothermia and dehydration in the treatment of severe cerebral hypoxia. Br J Anaesth. 1964;36:581–590.
195. et al. Experimental brain injury and delayed hypothermia. Surg Gynecol Obstet. 1960;110:27.
196. , . Bench-to-bedside review: hypothermia in traumatic brain injury. Crit Care. 2010;14(1):204.
197. et al. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2007;4(4).
198. et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346(8):557–563.
199. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346(8):549–556.
200. et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001;344(8):556–563.
201. et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001;345(1):66.
202. et al. Multicenter trial of early hypothermia in severe brain injury. J Neurotrauma. 2009;26(3):393–397.
203. et al. Hypothermia therapy after traumatic brain injury in children. N Engl J Med. 2008;358(23):2447–2456.
204. et al. Impact of hypotension and low cerebral perfusion pressure on outcomes in children treated with hypothermia therapy following severe traumatic brain injury: a post hoc analysis of the Hypothermia Pediatric Head Injury Trial. Dev Neurosci. 2010;32(5–6):406–412.
205. , , . Hypothermia for traumatic head injury. Cochrane Database Syst Rev. 2009;2.
206. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): temperature control. Pediatric Crit Care Med. 2012;13:S42–S45.
207. , , . Decompressive craniectomy and postoperative complication management in infants and toddlers with severe traumatic brain injuries. J Neurosurg Pediatr. 2009;3(4):334–339.
208. et al. Outcome following decompressive craniectomy in children with severe traumatic brain injury: a 10-year single-center experience with long-term follow up. J Neurosurg Pediatr. 2007;106(4):268–275.
209. , Sgouros S. Early decompressive craniectomy may be effective in the treatment of refractory intracranial hypertension after traumatic brain injury. Childs Nerv Syst. 2006;22(10):1268–1274.
210. et al. Decompressive craniectomy in pediatric patients with traumatic brain injury with intractable elevated intracranial pressure. J Pediatr Surg. 2006;41(1):83–87; discussion 83–87.
211. , , . Decompressive craniectomy for traumatic brain injury: when is it too late? Childs Nerv Syst. 2005;21(12):1014–1015.
212. et al. Severe traumatic brain injury in children—a single center experience regarding therapy and long-term outcome. Childs Nerv Syst. 2010;26(11):1563–1573.
213. et al. Early decompressive craniectomy and duraplasty for refractory intracranial hypertension in children: results of a pilot study. Crit Care. 2003;7(6):R133–R138.
214. et al. Decompressive craniectomy in children with nontraumatic refractory high intracranial pressure. J Neurosurg Pediatr. 2009;3(1):66–69.
215. . Decompressive craniectomy for the treatment of refractory high intracranial pressure in traumatic brain injury. Cochrane Database Syst Rev. 2009(1):CD003983.
216. . A randomized trial of very early decompressive craniectomy in children with traumatic brain injury and sustained intracranial hypertension. Childs Nerv Syst. 2001;17(3):154–162.
217. et al. Ventricular pressure monitoring during bilateral decompression with dural expansion. J Neurosurg. 1999;91(6):953–959.
218. et al. Intracranial pressure and cerebral oxygenation changes after decompressive craniectomy in a child with traumatic brain swelling. Childs Nerv Syst. 2007;23(11):1331–1335.
219. , , . A comparison of hinge craniotomy and decompressive craniectomy for the treatment of malignant intracranial hypertension: early clinical and radiographic analysis. Neurosurg Focus. 2009;26(6):E6.
220. et al. The surgical approach to the management of increased intracranial pressure after traumatic brain injury. Anesth Analg. 2010;111(3):736–748.
221. , , . Reversible monoparesis following decompressive hemicraniectomy for traumatic brain injury. J Neurosurg. 2008;109(2):245–254.
222. , . Assessment of outcome after severe brain damage: a practical scale. Lancet. 1975;305(7905):480–484.
223. et al. Decompressive Craniectomy in Diffuse Traumatic Brain Injury. N Engl J Med. 2011; 364:1493–1502.
224. . Clinical value of decompressive craniectomy. N Engl J Med. 2011;364(16):1558–1559.
225. et al. Is decompressive craniectomy useless in severe traumatic brain injury? Crit Care. 2011;15(5):193.
226. RESCUEicp Study 2011. http://www.rescueicp.com Accessed November 28, 2011.
227. et al. Decompressive craniectomy in traumatic brain injury: the randomized multicenter RESCUEicp study ( www.RESCUEicp.com). Brain Edema XIII. 2006:17–20.
228. et al. Update on the RESCUEicp decompressive craniectomy trial. Crit Care. 2011;15(suppl 1):312.
229. et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): decompressive craniectomy for the treatment of intracranial hypertension. Pediatr Crit Care Med. 2012;13:S53–S57.
230. , . Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17(5):367–388.
231. et al. The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J Neurotrauma. 2005;22(1):106–118.
232. et al. Effect of sex steroid hormones on brain edema, intracranial pressure, and neurologic outcomes after traumatic brain injury. Can J Physiol Pharmacol. 2010;88(4):414–421.
233. et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49(4):391–402, 402 e391–e392.
234. et al. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12(2):R61.
235. , , . Progesterone for acute traumatic brain injury. Cochrane Database Syst Rev. 2011(1):CD008409.
236. . Aquaporins: translating bench research to human disease. J Exp Biol. 2009;212(pt 11):1707–1715.
237. et al. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17(1):171–180.
238. et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6(2):159–163.
239. et al. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18(11):1291–1293.
240. et al. Modulation of AQP4 expression by the selective V1a receptor antagonist, SR49059, decreases trauma-induced brain edema. Acta Neurochir Suppl. 2008;102:425–429.
241. et al. Arginine-vasopressin V1a receptor inhibition improves neurologic outcomes following an intracerebral hemorrhagic brain injury. Neurochem Int. 2011;58(4):542–548.
242. , . Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci. 2005;6(6):484–494.
243. et al. Reduced functional deficits, neuroinflammation, and secondary tissue damage after treatment of stroke by nonerythropoietic erythropoietin derivatives. J Cereb Blood Flow Metab. 2007;27(3):552–563.
244. et al. Reduced brain edema and functional deficits after treatment of diffuse traumatic brain injury by carbamylated erythropoietin derivative. Crit Care Med. 2011;39(9):2099–2105.
245. et al. Safety and efficacy of erythropoietin in traumatic brain injury patients: a pilot randomized trial. Crit Care Res Pract. 2010;2010.
246. , , . The idolatry of the surrogate. Pediatr Crit Care Med. 2012;13:S7–S10.
247. Kochanek PM et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents (2nd edition): methods. Pediatr Crit Care Med. 2012;13:S7–S10.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


