Fig. 3.1
Schematic representation of the coagulation cascade. HK high-molecular-weight kininogen, PK prekallikrein, PL phospholipid, PT prothrombin, TH thrombin coagulation cascade
Fluid-phase coagulation components are involved in propagation of the clotting process, in termination of clotting by antithrombotic control mechanisms, and in the removal of the clot by fibrinolysis. At the same time, endothelium and peripheral blood monocytes play a main role in expressing TF and supporting clot formation, while platelets are pivotal in the initiation and formation of the plug.
aPL procoagulant mechanism(s) has been initially related to the antibody’s binding to β2GPI or PT and the consequent interference with the natural anticoagulant system of protein C and with the reduction of fibrinolysis. However, two main reasons supported the search for additional pathogenic mechanisms:
(a)
Vascular events in APS patients are affecting both arterial and venous vessels, while abnormal protein C and fibrinolysis may account mainly for venous events.
(b)
It is still not clear whether aPL react in a significant manner with the PL-binding proteins (β2GPI and PT) in fluid phase. In fact, all aPL are low-avidity antibodies suggesting that complex formation in the fluid phase requires stechiometric antigen-antibody ratios that are uncommon in patients [20].
As a consequence, a lot of investigations have been focused on aPL ability to react with PL-binding proteins expressed on the cell membrane of the cells involved in the coagulation cascade.
In this regard, β2GPI is expressed on the cell membranes at high antigenic density and so more easily recognized by low-avidity autoantibodies. In addition, it has been reported that circulating β2GPI is usually present in the circular-closed form, while it is opened only after binding to negative PL or after complexing with LPS. Since the autoantibodies were reported to react mainly with the opened form (i.e., reacting with the exposed cryptic epitope of DI), this finding may further explain the reason of the limited reactivity with the circulating form.
Most of the pathogenic mechanisms potentially responsible for thrombus formation have been demonstrated using in vitro models. However, three different in vivo models of thrombosis induced respectively by mechanical, chemical, or photochemical trauma have confirmed the pathogenic effect of aPL [2]. Two of these experimental models showed an increase of the thrombus size already triggered respectively by the mechanical or the chemical stimulus and a delayed dissolution. On the other hand, the third model showed that the passive infusion of human aPL IgG together with a small amount of LPS is able by itself to start clotting in the rat mesenteric arterial microcirculation [21].
Table 3.1 and Fig. 3.2 summarize the main pathogenic mechanisms that have been reported in the literature [2, 22].
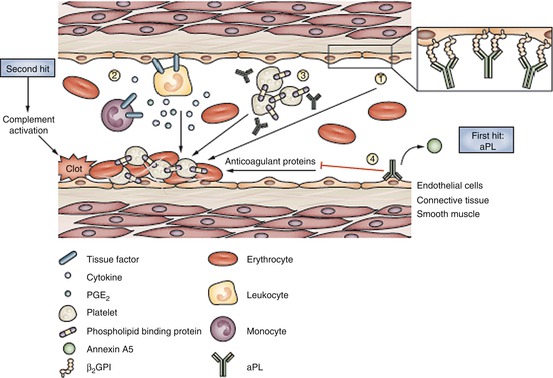
Table 3.1
Antiphospholipid antibody-mediated pathogenic mechanisms for thrombosis
Interference with fluid–phase coagulation components: |
Inhibition of natural anticoagulants |
Inhibition of protein C activationa |
Disruption of the annexin V shielda |
Inhibition of fibrinolysis |
Interference with cells of the coagulation cascade: |
Endothelial cell perturbationa |
Monocyte activation (TF expression)a |
Platelet activationa |
Complement activationa |
Fig. 3.2
Pathogenic clotting mechanisms mediated by aPL. aPL actions favor clot formation through several routes. 1 aPL interact with endothelial cells, primarily through binding of β2GPI on the cell surface, and induce a procoagulant and proinflammatory endothelial phenotype. 2 aPL upregulate tissue factor expression on endothelial cells and blood monocytes, and promote endothelial leukocyte adhesion, cytokine secretion, and PGE2synthesis. 3 aPL recognize phospholipid-binding proteins expressed on platelets—aPL binding potentiates platelet aggregation induced by another agonist. 4 aPL interfere with plasma components of the coagulation cascade, by inhibiting anticoagulant activity, by affecting fibrinolysis, and by displacing the binding of the natural anticoagulant annexin A5 to anionic structures. These mechanisms all contribute to a procoagulant state that is necessary but not sufficient for clotting. Clot formation seems to require two steps: the presence of aPL provides the ‘first hit’, which produces clotting when accompanied by another procoagulant condition, a ‘second hit’. Complement activation seems to be necessary for clot formation in vivo. Abbreviations: aPL anti-phospholipid autoantibodies, β2GPI β2 glycoprotein I, PGE2 prostaglandin E2 (Permission from Meroni et al [2])
Still open is the question why some aPL-positive patients develop both arterial and venous events and why others can display arterial or venous thrombosis only.
3.5 Interference of aPL with Fluid-Phase Coagulation Cascade
The evidence of aPL interference with fluid-phase components of coagulation has been provided mostly by in vitro models [2, 22].
aPL can bind some members of the serine protease (SP) family, which enlists proteins involved in hemostasis (procoagulant factors such as thrombin, PT, FVIIa, FIXa, and FXa and natural anticoagulants as protein C) and in fibrinolysis (plasmin and tissue plasminogen activator (tPA)). The fact that β2GPI and SP enzymatic domain share conformational epitopes was suggested to be the reason for the cross-reactivity.
aPL interact with thrombin and FXa, interfering with the formation of thrombin-antithrombin (AT) and FXa-AT complexes, thus affecting the AT inactivation of thrombin and FXa. Moreover, aPL are reported to decrease activated protein C (APC) activity by competing with APC for PL binding. Accordingly, an increased APC resistance has been reported in APS. Several groups have also found decreased levels of both proteins C and S and aPL with affinity for either protein S or C. In addition, some aPL were shown to inhibit plasmin-mediated fibrinolysis, eventually leading to an impairment of fibrin dissolution by plasmin. Some aPL subpopulations were also found to bind to tPA, inhibiting tPA-mediated conversion of plasminogen to plasmin. Anti-tPA antibodies were shown to inversely correlate with plasma tPA activity in APS.
Besides the inhibitory effects on the natural anticoagulant systems, aPL may also increase the enzymatic activity of procoagulants. In fact some aPL subpopulations were shown to induce a gain of function of PT leading to increased fibrin production. In addition, aPL have been reported to disrupt the crystallization of annexin A5 on EC monolayer. The “shield” of annexin A5 is thought to represent a potent anticoagulant barrier that prevents PL bioavailability for coagulation enzymes [2, 22].
3.6 Interference of aPL with Cells Involved in the Coagulation Cascade
More recently, research has focused on aPL interaction with cells involved in the hemostatic balance as platelets, monocytes, and EC [2].
3.6.1 Platelet Involvement
The occurrence of mild thrombocytopenia is a frequent clinical finding among APS patients and strongly suggested the possible interaction between platelets and aPL.
aPL reactivity with platelets has been shown only after their pre-activation by agonists such as thrombin or collagen. The reason for this was thought to be related to the exposure of phosphatidylserine (PS) on the cell outer membrane after platelet activation and the consequent binding of the cationic β2GPI. However, β2GPI can also bind two surface receptors on platelets. Apolipoprotein E receptor 2’ (ApoER2’) is a member of low-density lipoprotein (LDL) receptor family expressed by platelets; its LDL-binding domain I recognizes the PL-binding site of β2GPI domain V. Lastly β2GPI has been shown to bind directly GPIba, a subunit of the GPIb-IX-V platelet receptor [23] and to platelet factor 4 [24].
Altogether these findings suggest that the main antigenic target on platelets is the β2GPI itself which can be recognized by specific antibodies.
There is evidence that such interaction may be responsible for several biological effects. In general, the binding of the antibodies with their own antigenic target can trigger platelet activation. In addition, aPL have been shown to neutralize β2GPI interaction with von Willebrand factor (vWF). β2GPI acts as a biologically relevant inhibitor of vWF function, thus interfering with vWF-dependent platelet adhesion. This action could be hampered by aPL so favoring thrombosis and may explain the consumptive thrombocytopenia frequently observed in aPL carriers. Once bound, aPL have been shown to enhance the expression of platelet membrane glycoproteins (GPs) IIb/IIIa and IIIa. GPs are fibrinogen receptors that mediate platelet aggregation, whose role in APS pathogenesis is further supported by in vivo findings [25]. In fact, aPL did not affect thrombus formation in GPIIb/IIIa-deficient mice and pretreatment with a monoclonal anti-GPIIb/IIIa antibody inhibited aPL-mediated reduced thrombus formation. In vivo evidence of platelet activation by aPL has been gained in other models: (i) aPL produced a platelet-rich thrombus in rats, after treatment with low concentration of adenosine diphosphate (ADP), and (ii) platelet involvement was reported in the thrombus formation by photochemical injury in the rat. Platelet activation by aPL has been supported by ex vivo studies since elevated levels of thromboxane (TX, the major eicosanoid metabolic breakdown product) have been found in the urine of APS patients [26].
3.6.2 Involvement of Monocytes
aPL upregulate TF expression on peripheral blood monocytes. In particular, monocyte TF expression is increased in patients with APS and correlates with the expression of β2GPI. Accordingly, cell-surface TF on monocytes is higher in APS patients than in subjects without thrombotic events. Interestingly, vascular endothelial growth factor (VEGF) stimulates TF expression in monocytes through its tyrosine-kinase receptor Flt-1, and expression of both VEGF and Flt-1 is increased in monocytes from APS patients [27]. On monocytes, aPL have been suggested to interact with β2GPI in association with annexin A2 [28] and toll-like receptor (TLR) 4 within lipid rafts [29]. Other authors provided indirect evidence of TLR2 involvement in mediating aPL-induced monocyte activation [2].
3.6.3 Involvement of Endothelial Cells
Endothelium has been suggested to represent a cell type deeply involved in APS pathogenesis since the earliest pathogenic studies. The milestone was the demonstration that aPL could react with EC mainly recognizing β2GPI present on the cell membrane [30, 31]. Once bound the β2GPI-dependent aPL can trigger EC perturbation with the induction of a proinflammatory and procoagulant cell phenotype [2, 22, 26].
The majority of the studies have been performed in vitro showing an upregulation of endothelial cellular adhesion molecules (such as ICAM-1, VCAM-1, and E-selectin) and the synthesis of proinflammatory cytokines (as interleukin (IL)-1β, IL-6, and IL-8). Accordingly, in vivo studies have shown that passive infusion of aPL induced an increase in the rate of leukocytes adhering to the endothelium. Lastly, aPL may modulate the vascular tone by inhibiting endothelial nitric oxide synthase and altering prostaglandin metabolism [2, 26].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


