Chapter 32 Amenorrhea, Oligomenorrhea, and Hyperandrogenic Disorders∗
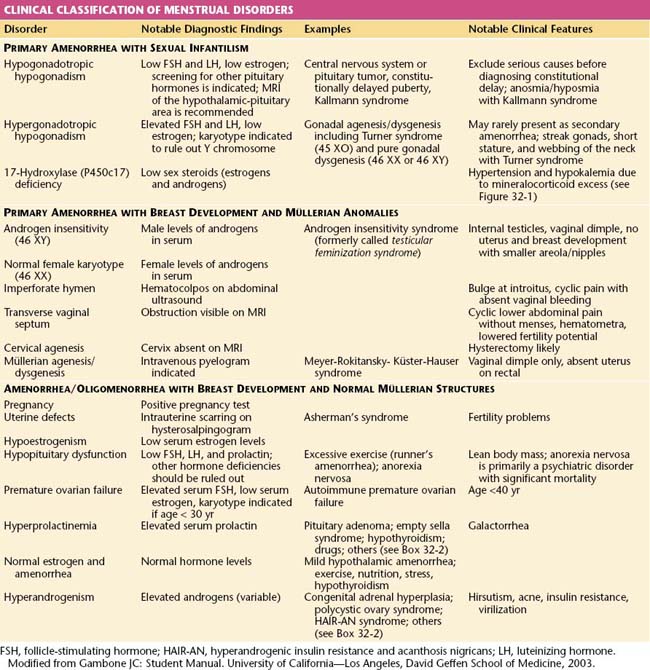
Amenorrhea, or the absence of menses, is a common symptom of several pathophysiologic states. This condition traditionally has been divided into primary amenorrhea, in which menarche (the first menses) has not occurred, and secondary amenorrhea, in which menses has been absent for 6 months or more. A more functional or clinical division of menstrual disorders based on initial history and physical examination would be as follows: primary amenorrhea with sexual infantilism, primary amenorrhea with breast development and müllerian anomalies, and amenorrhea and oligomenorrhea with breast development and normal müllerian structures. The last group includes disorders causing primary as well as secondary amenorrhea, oligomenorrhea, and the hyperandrogenic states (Table 32-1).
 Primary Amenorrhea
Primary Amenorrhea
PRIMARY AMENORRHEA WITH SEXUAL INFANTILISM
Hypergonadotropic Primary Amenorrhea and Sexual Infantilism
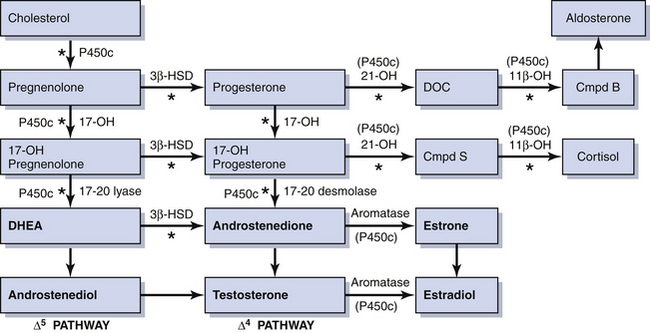
Rarely, some patients with primary amenorrhea and sexual infantilism have a defect of estrogen and androgen production. One example of this is a 17-hydroxylase (P450c17) deficiency, which prevents the synthesis of these sex steroids (Figure 32-1). These individuals have hypertension and hypokalemia caused by mineralocorticoid excess. Other patients, such as those with a 46 XY karyotype and Leydig cell agenesis, may lack the cells necessary for sex steroid production. Because the Leydig cells in the testicle are responsible for producing testosterone, these individuals are born with female external genitalia.
PRIMARY AMENORRHEA WITH BREAST DEVELOPMENT AND MÜLLERIAN ANOMALIES
Müllerian Dysgenesis or Agenesis
Patients with primary amenorrhea, breast development, and a 46 XX karyotype have levels of testosterone appropriate for females. This clinical diagnosis may be caused by müllerian defects that cause obstruction of the vaginal canal (e.g., imperforate hymen or a transverse vaginal septum) or by the absence of a normal cervix or uterus and normal fallopian tubes. An imperforate hymen should be suspected in adolescents who report monthly dysmenorrhea in the absence of vaginal bleeding (see Figure 18-7, pg 237). Clinically, these patients often present with a vaginal bulge and a midline cystic mass on rectal examination. Ultrasonography confirms the presence of a normal uterus and ovaries with a hematocolpos. These patients should be treated with hymenectomy.
Alternatively, women may present with similar symptoms but without a vaginal bulge. When ultrasonography confirms a normal uterus and ovaries, a transverse, obstructing vaginal septum (see Figure 18-8, pg 237) or cervical agenesis should be suspected. MRI is the diagnostic procedure of choice in these patients. If the MRI scan confirms a transverse septum, surgical correction is indicated. Surgical construction of a functional cervix is extremely difficult. In general, it is recommended that these women undergo hysterectomy.
 Amenorrhea and Oligomenorrhea with Breast Development and Normal Müllerian Structures
Amenorrhea and Oligomenorrhea with Breast Development and Normal Müllerian Structures
All patients with menstrual bleeding disorders should be tested for pregnancy. Once pregnancy has been excluded, these individuals can be characterized as shown in Table 32-1. Initial history taking should include questions about the timing of thelarche, pubarche, and menarche. The timing and development of the menstrual disorder (present since puberty or new), significant weight change, strenuous exercise activities, dietary habits, sexual activity, concomitant illnesses or complaints, abnormal facial or body hair growth, scalp hair loss, acne, and the presence or absence of hot flashes and vaginal dryness should be noted. A comprehensive list of medications and dietary supplements taken should be obtained.
AMENORRHEA AND OLIGOMENORRHEA ASSOCIATED WITH HYPOESTROGENISM
Premature Ovarian Failure
Premature ovarian failure is defined as ovarian failure before the age of 40 years (see Chapter 35). When it occurs in patients younger than 30 years of age, ovarian failure may be caused by a chromosomal disorder. A karyotype is performed to exclude mosaicism (i.e., some cells bearing a Y chromosome). If cells with a Y chromosome are present, a gonadectomy to prevent malignant transformation is indicated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


