Wendy J. Schillings
Howard D. McClamrock
A complex hormonal interaction must take place in order for normal menstruation to occur. The hypothalamus must secrete gonadotropin-releasing hormone (GnRH) in a pulsatile fashion, which is modulated by neurotransmitters and hormones. The GnRH stimulates secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the pituitary, which promotes ovarian follicular development and ovulation. A normally functioning ovarian follicle secretes estrogen; after ovulation, the follicle is converted to corpus luteum, and progesterone is secreted in addition to estrogen. These hormones stimulate endometrial development. If pregnancy does not occur, estrogen and progesterone secretion decrease and withdrawal bleeding begins. If any of the components (hypothalamus, pituitary, ovary, uterus, and outflow tract) are nonfunctional, bleeding cannot occur.
The mean age of menarche became younger during this century. Therefore, the definition of primary amenorrhea changed: Primary amenorrhea is defined as the absence of menses by 13 years of age when there is no visible development of secondary sexual characteristics or by 15 years of age in the presence of normal secondary sexual characteristics. The ages defining primary amenorrhea were decreased by 1 year to continue to represent two standard deviations above the mean age of developing secondary sexual characteristics and menses (1). Failure to begin breast development by age 13 warrants investigation. A woman who previously menstruated can develop secondary amenorrhea, which is defined as absence of menstruation for three normal menstrual cycles (2). A woman with regular cycles and a delay of menses of even a week may warrant assessment with a pregnancy test. It is reasonable to evaluate a woman who has fewer than nine cycles per year. With a few exceptions, the causes of primary amenorrhea are similar to the causes of secondary amenorrhea.
Patients may develop slight alterations in the hypothalamic–pituitary–ovarian axis that are not severe enough to cause amenorrhea but instead cause irregular menses (oligomenorrhea) associated with absent or infrequent ovulation. These patients may bleed excessively during menstruation because estrogen is unopposed. The etiologies of oligomenorrhea overlap with the etiologies of amenorrhea, with the exception that certain anatomic (e.g., absent uterine development) and karyotypic abnormalities (e.g., Turner syndrome), are largely associated with primary amenorrhea.
The World Health Organization (WHO) described three classes of amenorrhea. WHO Group I includes women with no evidence of endogenous estrogen production, normal or low FSH levels, normal prolactin levels, and no lesion in the hypothalamic-pituitary region. WHO Group II is associated with evidence of estrogen production and normal levels of prolactin and FSH. WHO Group III includes individuals with elevated serum FSH indicating gonadal insufficiency or failure.
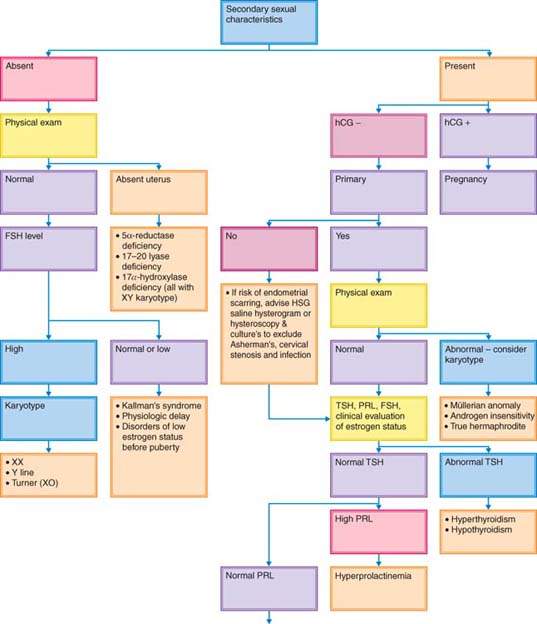
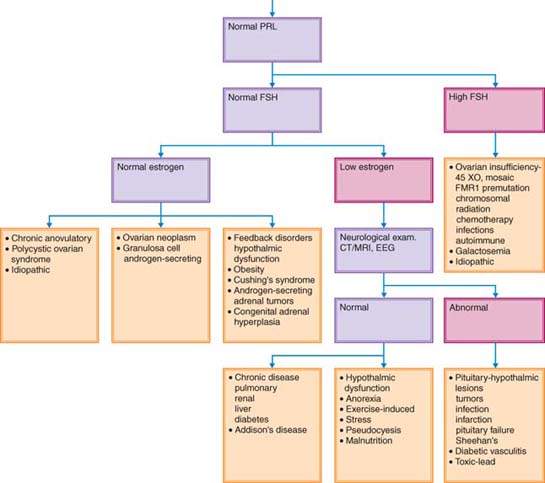
To detect the cause of amenorrhea, it is useful to determine whether secondary sexual characteristics are present (Fig. 30.1). The absence of secondary sexual characteristics indicates that a woman was never exposed to estrogen.
Figure 30.1 Decision tree for evaluation of amenorrhea. FSH, follicle-stimulating hormone; HCG, human chorionic gonadotropin; HSG, hysterosalpingogram; TSH, thyroid-stimulating hormone; PRL, prolactin; CT, computed tomography; MRI, magnetic resonance imaging; EEG, electroencephalogram; SHG, saline hysterogram.
Amenorrhea without Secondary Sexual Characteristics
Although the diagnosis and treatment of disorders associated with hypogonadism were discussed in another chapter (see Chapter 29), they will be mentioned here because these conditions may present as primary amenorrhea. Because breast development is the first sign of estrogen exposure in puberty, patients without secondary sexual characteristics typically have primary, not secondary, amenorrhea (Fig 30.1). It is helpful to categorize the causes of amenorrhea in the absence of breast development on the basis of gonadotropin status.
Causes of Primary Amenorrhea
Hypergonadotropic Hypogonadism Associated with Absence of Secondary Sexual Characteristics
Gonadal dysgenesis is a term typically used to describe abnormal development of the gonads, typically resulting in streak gonads. Gonadal dysgenesis is associated with high levels of LH and FSH because the gonad fails to produce the steroids and inhibin that would normally feed back to the pituitary gland to suppress pituitary production of LH and FSH. Karyotypic abnormalities are common in women with primary amenorrhea associated with gonadal failure (Table 30.1). In one series, approximately 30% of patients with primary amenorrhea had an associated karyotypic abnormality (3). Turner syndrome (45,X) and its variants represent the most common form of hypergonadotropic hypogonadism in women with primary amenorrhea. Other disorders associated with primary amenorrhea include structurally abnormal X chromosomes, mosaicism (e.g., 45,X in some cells and another karyotype such as 46,XX or 46,XXX in other cells), pure gonadal dysgenesis (46,XX and 46,XY individuals with gonadal streaks resulting from lack of gonad development), enzyme deficiencies that prevent normal estrogen production, and gonadotropin-receptor inactivating mutations. Individuals with these conditions have gonadal failure and cannot synthesize ovarian steroids. Therefore, gonadotropin levels are elevated because of the lack of negative estrogen feedback on the hypothalamic–pituitary axis. Most patients with these conditions have primary amenorrhea and lack secondary sexual characteristics. Occasionally patients with a partial deletion of the X chromosome, mosaicism, or pure gonadal dysgenesis (46,XX) may synthesize enough estrogen in early puberty to induce breast development and a few episodes of uterine bleeding and thus have secondary amenorrhea. Ovulation and, occasionally, pregnancy are possible.
Table 30.1 Amenorrhea Associated with a Lack of Secondary Sexual Characteristics
| Abnormal pelvic examination |
| 5α-reductase deficiency, 17, 20-lyase deficiency, or 17α-hydroxylase deficiency in |
| XY individual |
| Congenital lipoid adrenal hyperplasia |
| Luteinizing hormone receptor defect |
| Hypergonadotropic hypogonadism |
| Gonadal dysgenesis |
| Follicle-stimulating hormone receptor defect |
| Partial deletion of X chromosome |
| Sex chromosome mosaicism |
| Environmental and therapeutic ovarian toxins |
| 17α-hydroxylase deficiency in XX individual |
| Galactosemia |
| Congenital lipoid adrenal hyperplasia in XX individual |
| Hypogonadotropic hypogonadism |
| Physiologic delay |
| Kallmann syndrome |
| Central nervous system tumors |
| Hypothalamic/pituitary dysfunction |
Genetic Disorders
Turner Syndrome
Turner syndrome (45,X) is the most common karyotypic abnormality causing gonadal failure and primary amenorrhea (3,4). It appears that patients with Turner syndrome initially have normal ovarian development in utero. Amenorrhea is the result of accelerated atresia of the follicles. The fibrotic ovaries are called streak ovaries.
In addition to gonadal failure, there are associated stigmata with Turner syndrome that include short stature, webbed neck, shield chest, cubitus valgus (increased carrying angle of the arms), low hair line, high arched palate, multiple pigmented nevi, and short fourth metacarpals (4). X inactivation is a process that inactivates most of the genes on one X chromosome. Of the genes on the X chromosome, 20% escape X inactivation, and it is believed that loss of the second copy of these genes in a 45,X patient causes the stigmata associated with Turner syndrome (5).
After the diagnosis of Turner syndrome is confirmed by karyotype, studies should be performed to ensure that cardiac (30% have coarctation of the aorta), renal (especially horseshoe kidney), and autoimmune (thyroiditis) abnormalities are diagnosed and treated. Cardiac magnetic resonance imaging (MRI) should be used in addition to echocardiography (6). Evaluation should be performed in childhood to identify potential attention-deficit or nonverbal learning disorders. Women with Turner syndrome should be screened for diabetes mellitus, aortic enlargement, hypertension, and hearing loss throughout their lives (6).
Abnormal X Chromosome
Those 46,XX individuals with partial deletions of the X chromosome have variable phenotypes depending on the amount and location of the missing genetic material. Patients with a deletion of the long arm of the X chromosome (Xq−) from Xq13 to Xq26 have sexual infantilism, normal stature, no somatic abnormalities, and streak gonads (7). Some patients may be eunuchoid in appearance and have delayed epiphyseal closure. Patients with a deletion of the short arm of the X chromosome (Xp) usually are phenotypically similar to individuals with Turner syndrome (8). Many genes on the Xp chromosome escape X inactivation and act similarly to genes on autosomes. The effective monosomy created by the deletion results in the phenotypic features of Turner syndrome (5). Most patients with a ring X have ovarian failure and phenotypes similar to Turner syndrome, although some are able to reproduce successfully. These patients differ from those with Turner syndrome in that they are more likely to have intellectual disability and have syndactyly. Patients with isochrome of the long arm of the X chromosome (i[Xq]) are similar to XO patients, with the exception that autoimmune disorders are more common. Half of the women with balanced translocations of the X chromosome to an autosome have gonadal failure. Typically, the normal X is inactivated to preserve the balance of autosomal genes. The gonadal failure can be caused by the chromosomal break occurring in a gene that is required for ovarian function, abnormal meiosis, or X inactivation of the translocated X and adjacent autosomal genes (5,9).
Mosaicism
Primary amenorrhea is associated with various mosaic states, the most common of which is 45,X/46,XX (10). The clinical findings in 45,X/47,XXX and 45,X/46,XX/47,XXX are similar to those in 45,X/46,XX and vary in estrogen and gonadotropin production, depending on the number of follicles in the gonads. When compared with the pure 45,X cell line, individuals with 45,X/46,XX are taller and have fewer abnormalities, although 80% of those with 45,X/46,XX mosaics are shorter than their peers, and 66% have some somatic abnormalities. Spontaneous menstruation occurs in approximately 20% of these patients (10).
Pure Gonadal Dysgenesis
Individuals who are phenotypically female with sexual infantilism, primary amenorrhea, normal stature, and no karyotypic abnormalities (46,XX or 46,XY) have pure gonadal dysgenesis. The gonads are usually streaks, but there may be some development of secondary sexual characteristics, and a few episodes of uterine bleeding. Pure gonadal dysgenesis in a 46,XY individual (previously known as Swyer syndrome) can occur when mutations in the SRY (sex-determining region gene on the Y chromosome) located at Yp11 result in XY females without proper gonad development (11,12).
Mutations in many other genes such as SOX9, DAX1, WT-1, and SF1, which affect testicular differentiation and inhibit antimüllerian hormone production, result in XY pure gonadal dysgenesis (13). The SOX9 gene located at 17q24 has a role in testis differentiation and promotes antimüllerian hormone secretion. Some but not all mutations in the SOX9 gene cause XY sex reversal, along with camptomelic dysplasia (severe skeletal abnormalities) (14,15). Duplications of the DAX1 gene at Xp21 cause dose-sensitive XY sex reversal (16). DAX1 is hypothesized to antagonize the SRY gene, preventing testis development. Transgenic XY mice with overexpression of the DAX1 gene develop as phenotypic females, supporting this hypothesis (17). Mutation in the WT1 gene (Wilms’ tumor suppressor gene 1) located at 11p13 causes several different syndromes, depending on where the mutation in the gene occurs. In Frasier syndrome, there is alternative splicing, which causes the protein product to lack a highly conserved KTS triplet repeat. The normal +KTS isoform of the protein is believed to synergize with SF1 (steroidogenic factor 1) to promote the expression of antimüllerian hormone (AMH). Lack of the +KTS isoform in an XY patient results in normal female internal and external genitalia, streak gonads, and progressive glomerulopathy. These women frequently develop gonadoblastomas but rarely develop Wilms’ tumor, which is associated with mutations in other locations in the WT1 gene. XX patients with the mutation that prevents the +KTS isoform have similar kidney abnormalities but develop normal ovaries and genitalia (17–19). One XY patient with a heterozygous mutation of the SF1 gene had adrenal failure and sex reversal (20). SF1 is an orphan nuclear receptor that regulates AMH expression and regulates all the cytochrome P450 steroid hydroxylase enzymes (19). Duplication of 1p, which encodes the WNT4 gene, causes XY sex reversal. WNT4 may upregulate DAX1 transcripts. A TRX mutation causes XY sex reversal. Other genes that cause XY gonadal dysgenesis are likely to be identified. Mutations at 9p24 and 10q cause XY sex reversal, but the exact genes causing the defects are not elucidated (17,19,21).
XX pure gonadal dysgenesis can be caused by the presence of small Y chromosome fragments in the genome. It is estimated that 5% to 40% of patients with Ullrich-Turner syndrome have Y sequences by polymerase chain reaction (PCR), depending on the DNA sequences targeted for testing (22,23). If Y sequences are present, gonadectomy is advised because of the risk of gonadoblastoma (22).
In other patients with XX gonadal dysgenesis, the condition is likely to be caused by gene mutations that lead to ovarian insufficiency before pubertal development or after the development of secondary sexual characteristics, as discussed later in this chapter.
Mixed Gonadal Dysgenesis
Most patients with mixed gonadal dysgenesis are XY and have ambiguous genitalia with a streak gonad on one side and a malformed testis on the opposite. A small proportion of these patients have mutations in the SRY gene (17).
Rare Enzyme Deficiencies
Congenital Lipoid Adrenal Hyperplasia
Patients with this autosomal recessive disorder are unable to convert cholesterol to pregnenolone, which is the first step in steroid hormone biosynthesis. A defect was not found in the P450scc gene, which is the conversion enzyme responsible for this step in the pathway. Instead, 15 different mutations were identified in the steroidogenic acute regulatory protein (StAR), which facilitates the transport of cholesterol from the outer to the inner mitochondrial membrane. This protein appears to be the rate-limiting step for steroid hormone biosynthesis stimulated by tropic hormones. These patients present in infancy with hyponatremia, hyperkalemia, and acidosis. Both XX and XY individuals are phenotypically female. Genetic clusters of the disorder are found in the Japanese, Korean, and Palestinian Arab populations. With appropriate mineralocorticoid and glucocorticoid replacement, these patients can survive into adulthood. Most patients are XY and do not have a uterus. Without hormone replacement, they remain sexually infantile. XX patients may acquire secondary sexual characteristics at puberty but develop large ovarian cysts and early ovarian failure (24,25).
17á-Hydroxylase and 17,20-lyase Deficiency
Mutations in the CYP17 gene cause abnormalities in both the 17α–hydroxylase and 17,20-lyase functions of the protein that is active in the adrenal and gonadal steroidogenic pathways. More than 20 mutations that alter the reading frame of the gene are identified, even though very few people have the disorder (26). Patients have either 46,XX or 46,XY karyotypes. The uterus is absent in individuals with 46,XY karyotype, a feature distinguishing them from individuals with the 46,XX karyotype. Individuals with CYP17 mutations have primary amenorrhea, no secondary sexual characteristics, female phenotype, hypertension, and hypokalemia (27). The diminished levels of 17α-hydroxylase that characterize this disorder lead to a reduction in cortisol production, which in turn causes an increase in adrenocorticotropic hormone (ACTH). 17α-hydroxylase is not required for production of mineralocorticoids; thus, excessive amounts of mineralocorticoid are produced, resulting in sodium retention, loss of potassium, and hypertension. Patients with 17α–hydroxylase deficiency have primordial follicles, but gonadotropin levels are elevated because the enzyme deficiency prevents synthesis of sex steroids.
Aromatase Deficiency
This very rare autosomal recessive abnormality prevents the affected individual from aromatizing androgens to estrogen (28). This syndrome may be suspected even before birth because most mothers of affected children become virilized during pregnancy. This occurs because the placenta cannot convert the fetal androgens to estrogen and they diffuse into the maternal circulation. At birth, a female child has clitoromegaly and posterior labioscrotal fusion (ambiguous genitalia). At puberty, there is no breast development, primary amenorrhea, worsening virilization, absent growth spurt, delayed bone age, and multicystic ovaries. The diagnostic hormonal pattern consists of an elevation of FSH, LH, testosterone, and dehydroepiandrosterone sulfate (DHEAS) levels, and undetectable levels of estradiol. Estrogen therapy improves the ovarian and skeletal abnormalities but must be titrated to mimic normal estrogen levels. Estrogen administration should be minimal during childhood and increased at puberty (29,30).
Galactosemia
In girls, galactosemia often is associated with ovarian failure, but this condition usually is detected by newborn screening programs. A galactose-1 phosphate uridyl transferase level can be measured to assess the patient for galactosemia or carrier status.
Rare Gonadotropin Receptor Mutations
Luteinizing Hormone Receptor Mutation
Inactivation of LH receptors is identified in XY pseudohermaphrodites with primary amenorrhea in the absence of secondary sexual characteristics caused by homozygous premature stop codon, deletions, and missense mutations in the LHR gene located on chromosome 2. The Leydig cells in these individuals are unable to respond to LH, causing Leydig cell hypoplasia. This leads to early testicular failure and prevents masculinization. XX siblings with the same mutations develop normal secondary sexual characteristics but are amenorrheic with elevated LH levels, normal FSH levels, and cystic ovaries (31,32).
Follicle-Stimulating Hormone Receptor Mutation
An autosomal recessive single amino acid substitution in the extracellular domain of the FSH receptor, which prevents FSH binding, was identified in six families in Finland. This condition leads to primary or early secondary amenorrhea, variable development of secondary sexual characteristics, and high levels of FSH and LH (33).
Other Causes of Primary Ovarian Failure without Secondary Sexual Characteristics
Severe damage to the ovaries before the onset of puberty can lead to ovarian insufficiency and failure to develop secondary sexual characteristics. Ovarian dysfunction can occur in association with irradiation of the ovaries, chemotherapy with alkylating agents (e.g., cyclophosphamide), or combinations of radiation and other chemotherapeutic agents (34,35). Other causes of premature ovarian failure (also known as primary ovarian insufficiency) are more commonly associated with amenorrhea after the development of secondary sexual characteristics, as described below.
Hypogonadotropic Hypogonadism Associated with the Absence of Secondary Sex Characteristics
Primary amenorrhea resulting from hypogonadotropic hypogonadism occurs when the hypothalamus fails to secrete adequate amounts of GnRH or when a pituitary disorder associated with inadequate production or release of pituitary gonadotropins is present.
Physiologic Delay
Physiologic or constitutional delay of puberty is the most common manifestation of hypogonadotropic hypogonadism. Amenorrhea may result from the lack of physical development caused by delayed reactivation of the GnRH pulse generator. Levels of GnRH are functionally deficient in relation to chronologic age but normal in terms of physiologic development.
Kallmann Syndrome
The second most common hypothalamic cause of primary amenorrhea associated with hypogonadotropic hypogonadism is insufficient pulsatile secretion of GnRH (Kallmann syndrome), which has varied modes of genetic transmission. Insufficient pulsatile secretion of GnRH leads to deficiencies in FSH and LH (36). Kallmann syndrome is often associated with anosmia (inability to perceive odors), although a woman may not be aware of her impaired sense of smell. The hypogonadism and anosmia arise because of failure of proper neuronal migration during fetal development.
Other Causes of Gonadotropin-Releasing Hormone Deficiency
Deficiencies in GnRH may be caused by developmental or genetic defects, inflammatory processes, tumors, vascular lesions, or trauma. Central nervous system tumors that lead to primary amenorrhea, the most common of which is craniopharyngioma, are usually extracellular masses that interfere with the synthesis and secretion of GnRH or stimulation of pituitary gonadotropins. Virtually all of these patients have disorders in the production of other pituitary hormones and LH and FSH (37,38). Prolactin-secreting pituitary adenomas are rare in childhood and more commonly occur after development of secondary sexual characteristics.
Genetic Disorders
5α-Reductase Deficiency
5α-Reductase deficiency should be considered a cause of amenorrhea (39). Patients with this disorder are genotypically XY, frequently experience virilization at puberty, have testes (because of functioning Y chromosomes), and have no müllerian structures as a result of functioning AMH. 5α-Reductase converts testosterone to its more potent form, dihydrotestosterone. Patients with 5α–reductase deficiency differ from patients with androgen insensitivity because they do not develop breasts at puberty (Fig. 30.2). These patients have low gonadotropin levels as a result of testosterone levels that are sufficient to suppress breast development and allow normal feedback mechanisms to remain intact. Normal male differentiation of the urogenital sinus and external genitalia do not occur because dihydrotestosterone is required for this development. Normal internal male genitalia derived from the wolffian ducts are present because this development requires only testosterone. Male pattern hair growth, muscle mass, and voice deepening are testosterone dependent.
Gonadotropin-Releasing Hormone Receptor Mutations
Several mutations are identified in the GnRH receptor gene that causes abnormal GnRH function. Most affected patients are compound heterozygotes, but homozygous autosomal recessive mutations are identified. The GnRH receptor is a G-protein–coupled receptor. Functional studies show that the mutations cause marked decrease in binding of GnRH to its receptor or prevent second-messenger signal transduction. Without a functional signal transduction, FSH and LH are not stimulated and are unable to promote follicular growth (40). All patients are normosomic. Receptor mutations in GnRH cause 17% of sporadic cases of idiopathic hypogonadotropic hypogonadism with normal olfaction (41).
Follicle-Stimulating Hormone Deficiency
Patients with FSH deficiency usually seek treatment for delayed puberty and primary amenorrhea associated with hypoestrogenism. They are distinguished from other hypoestrogenic patients by having decreased FSH levels and increased LH levels. These patients have low serum androgen levels despite the abnormal LH-to-FSH ratio, indicating that FSH-stimulated follicular development is a prerequisite for thecal cell androgen production. In some of these patients, autosomal recessive mutations in the FSHβ subunit, which impair dimerization of α and β subunits and prevent binding to the FSH receptor, are identified (42). Pregnancy was achieved in one patient after induction of ovulation with injectable gonadotropins (43).
Other Hypothalamic/ Pituitary Dysfunctions
Functional gonadotropin deficiency results from malnutrition, malabsorption, weight loss or anorexia nervosa, excessive exercise, chronic disease, neoplasias, and marijuana use, although these conditions are more commonly associated with amenorrhea accompanied by secondary sexual characteristics that developed before the onset of the problem, which is discussed in detail below (44–48). Hypothyroidism, polycystic ovarian syndrome (PCOS), Cushing syndrome, hyperprolactinemia, and infiltrative disorders of the central nervous system are more commonly associated with amenorrhea in the presence of development of secondary sexual characteristics, but can lead to amenorrhea accompanied by delayed puberty (49,50). Constitutional delay without underlying causes is less common in girls than in boys, and the reason for lack of development should be vigorously pursued (51).
Evaluation of Women with Amenorrhea Associated with the Absence of Secondary Sexual Characteristics
A careful history and physical examination are necessary to appropriately diagnose and treat primary amenorrhea associated with hypogonadism. The physical examination may be particularly helpful in patients with Turner syndrome. A history of short stature but consistent growth rate, a family history of delayed puberty, and normal physical findings (including assessment of smell, optic discs, and visual fields) may suggest physiologic delay. Headaches, visual disturbances, short stature, symptoms of diabetes insipidus, and weakness of one or more limbs suggest central nervous system lesions (38). Galactorrhea may be seen with prolactinomas, a condition more commonly associated with secondary amenorrhea in the presence of normal secondary sexual characteristics.
The diagnostic workup is summarized as follows:
Treatment of Amenorrhea Associated with the Absence of Secondary Sexual Characteristics
Individuals with primary amenorrhea associated with all forms of gonadal failure and hypergonadotropic hypogonadism need cyclic estrogen and progestogen therapy to initiate, mature, and maintain secondary sexual characteristics. Prevention of osteoporosis is an additional benefit of estrogen therapy:
If possible, therapeutic measures are aimed at correcting the primary cause of amenorrhea:
Individuals whose karyotypes contain a Y cell line (45,X/46,XY mosaicism, or pure gonadal dysgenesis 46,XY) are predisposed to gonadal ridge tumors, such as gonadoblastomas, dysgerminomas, and yolk sac tumors. The gonads of these individuals should be removed when the condition is diagnosed to prevent malignant transformation. There is some evidence that hirsute individuals without Y chromosomes should undergo gonad removal. One patient with hirsutism and the karyotype 45,X was noted to have a streak gonad; the contralateral gonad was dysgenic and contained developing follicles, well-differentiated seminiferous tubules, and Leydig cells. This patient was found to be HY antigen–positive (57).
Clomiphene citrate is most often ineffective for inducing ovulation in patients with hypogonadism who desire pregnancy because such patients are hypoestrogenic. In patients with hypogonadism, ovulation induction with injectable gonadotropins is generally successful. In patients without ovarian function, oocyte donation may be appropriate. There are reports of deaths in pregnant patients with Turner syndrome resulting from aortic dissection and rupture (58). Careful counseling and investigation should be undertaken in patients with Turner syndrome before treating them with donated oocytes.
Amenorrhea with Secondary Sexual Characteristics and Abnormalities of Pelvic Anatomy
Causes
Outflow and Müllerian Anomalies
Amenorrhea occurs if there is blockage of the outflow tract, if the outflow tract is missing, or if there is no functioning uterus (Table 30.2) In order for menses to occur, the endometrium must be functional and there must be patency of the cervix and vagina. Most women with müllerian abnormalities will have normal ovarian function and thus will have normal secondary sexual characteristic development.
Table 30.2 Anatomic Causes of Amenorrhea
| Secondary sexual characteristics present |
| Müllerian anomalies |
| Imperforate hymen |
| Transverse vaginal septum |
| Mayer-Rokitansky-Küster-Hauser syndrome |
| Androgen insensitivity |
| True hermaphrodism |
| Absent endometrium |
| Asherman syndrome |
| Secondary to prior uterine or cervical surgery |
| Curettage, especially postpartum |
| Cone biopsy |
| Loop electroexcision procedure |
| Secondary to infections |
| Pelvic inflammatory disease |
| Intrauterine devise–related |
| Tuberculosis |
| Schistosomiasis |
Transverse Blockages
Any transverse blockage of the müllerian system will cause amenorrhea (59). Such outflow obstructions include imperforate hymen, transverse vaginal septum, and absence of the cervix or vagina. Transverse blockage of the outflow tract with an intact endometrium frequently causes cyclic pain without menstrual bleeding in adolescents. The blockage of blood flow can cause hematocolpos, hematometra, or hemoperitoneum, and endometriosis.
Müllerian Anomalies
Mayer-Rokitansky-Küster-Hauser syndrome includes vaginal agenesis with variable müllerian duct abnormalities accompanied in some cases by renal, skeletal, and auditory abnormalities (60). Müllerian agenesis accounts for approximately 10% of cases of primary amenorrhea (2). Of the patients with this syndrome, 15% have an absent, pelvic, or horseshoe kidney, 40% have a double urinary collecting system, and 5% to 12% have skeletal abnormalities (61–63). Mayer-Rokitansky-Küster-Hauser syndrome is associated with abnormal galactose metabolism (64).
Absence of Functioning Endometrium
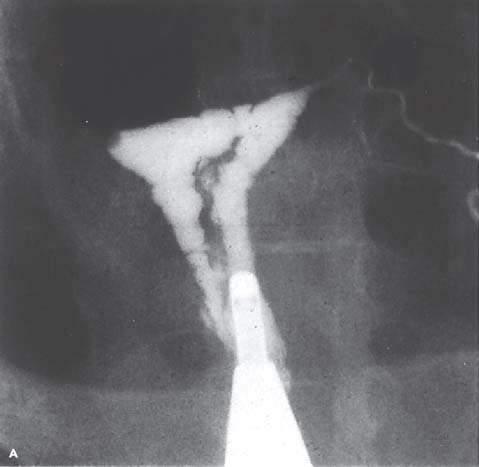
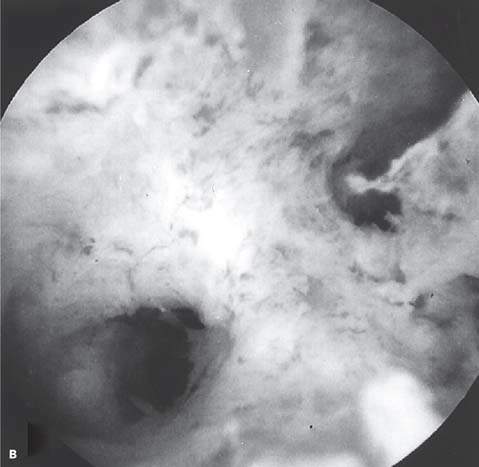
Amenorrhea may occur if there is no functioning endometrium. When the findings of the physical examination are normal, anatomic abnormalities of the uterine cavity should be considered. A congenitally absent endometrium is a rare finding in patients with primary amenorrhea. Asherman syndrome, which is more common with secondary amenorrhea or hypomenorrhea, may occur in patients with risk factors for endometrial or cervical scarring (Fig. 30.3). Such risk factors include a history of uterine or cervical surgery, infections related to use of an intrauterine device, and severe pelvic inflammatory disease. Asherman syndrome is found in 39% of patients undergoing hysterosalpingography who previously underwent postpartum curettage (65). Infections such as tuberculosis and schistosomiasis may cause Asherman syndrome but are not common for women who have lived their whole lives in the United States. Cervical stenosis resulting from surgical removal of dysplasia (cone biopsy, loop electroexcision procedure) may lead to amenorrhea.
Figure 30.2 A: A well-developed patient with complete androgen insensitivity. Note the characteristic paucity of pubic hair and well-developed breasts. (From Yen SSC, Jaffe RB. Reproductive endocrinology. 3rd ed. Philadelphia, PA: WB Saunders, 1991:497, with permission.) B: Another patient with androgen insensitivity syndrome with a contrasting thin body hiatus. This is a 17-year-old twin 46,XY. (From Jones HW Jr, Scott WW. Hermaphrodism, genital anomalies, and related endocrine disorders. 2nd ed. Baltimore, MD: Williams & Wilkins, 1971, with permission.)
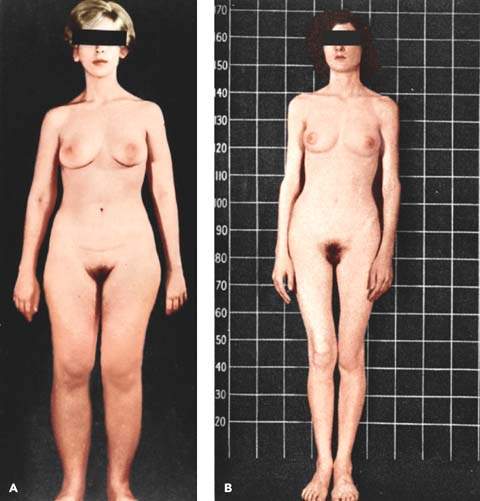
Figure 30.3 A: Intrauterine adhesion seen on hysterosalpingogram in a patient with Asherman syndrome. (From Donnez J, Nisolle M. The encyclopedia of visual medicine series—an atlas of laser operative laparoscopy and hysteroscopy. New York: Parthenon, 1994:306, with permission.) B: Hysteroscopic view of intrauterine adhesion in a patient with Asherman syndrome.
Androgen Insensitivity
Phenotypic females with complete congenital androgen insensitivity (previously called testicular feminization) develop secondary sexual characteristics but do not have menses (Fig. 30.2). Genotypically, they are male (XY) but have a defect that prevents normal androgen receptor function, leading to the development of the female phenotype. Serum testosterone is in the normal male range. The vagina may be absent or short.
Defects in the androgen receptor gene located on the X chromosome include absence of the gene that encodes for the androgen receptor and abnormalities in the binding domains of the receptor. Androgen receptor deficits are diverse and may result from diminished receptor function or concentration. The diversity of androgen receptor mutations may be related to diversity in phenotype. More than 250 extremely diverse mutations are described, with amino acid substitution being by far the most common (66,67). Postreceptor defects can exist (68). Total serum testosterone concentration is in the range of normal males. Because antimüllerian hormone is present and functions normally in these patients, internal female (müllerian) structures such as a uterus, vagina, and fallopian tubes are absent. Testes rather than ovaries are present in the abdomen or in inguinal hernias because of the presence of normally functioning genes on the Y chromosome. Patients have a blind vaginal pouch and scant or absent axillary and pubic hair. These patients experience abundant breast development at puberty; however, the nipples are immature and the areolae are pale. Testosterone is not present during development to suppress the formation of breast tissues; at puberty, the conversion of testosterone to estrogen stimulates breast growth. Patients are unusually tall with eunuchoidal tendency (long arms with big hands and feet).
True Hermaphroditism
True hermaphroditism is a rare condition that should be considered as a possible cause of amenorrhea. Both male and female gonadal tissues are present in these patients, in whom XX, XY, and mosaic genotypes are found. Two-thirds of the patients menstruate, but menstruation was never reported in XY genotypes. The external genitalia usually are ambiguous, and breast development frequently occurs in these individuals. Fifteen percent of XX true hermaphrodites have SRY translocations, and another 10% have Y chromosomal mosaicism within the gonad (17).
Evaluation of Women with Amenorrhea, Normal Secondary Sexual Characteristics, and Suspected Anatomic Abnormalities
Most congenital abnormalities can be diagnosed by physical examination:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


