This chapter discusses IEMs which produce AME from the perspective of an intensivist and a pediatric neurologist caring for a patient with AME for whom routine biochemical test results are available (basic metabolic panel, ammonia, blood gases, urine analysis). It provides guidance about when to suspect and how to diagnose and treat IEMs. I summarize the general classification of IEMs and discuss the initial approach to them. I then explain when to suspect these disorders and provide a synopsis of some conditions with emphasis on clinical presentation, biochemical findings, and treatment. More extended descriptions of many of the individual disorders mentioned in this chapter can be found in other textbooks cited in the reference notes (1,2).
Which Classes of IEMs Sometimes Produce AME as Symptoms?
From a pathophysiologic perspective, IEMs can be classified into 3 groups (1):
Group 1—complex molecule: This group of disorders is caused by defects in the synthesis or catabolism of complex molecules. Symptoms of these disorders are progressive and permanent and are independent of any intercurrent illnesses or food intake. Lysosomal storage disorders (such as mucopolysaccharidosis, Gaucher, Fabry), peroxisomal biogenesis defects, and congenital disorders of glycosylation (CDG) belong to this group. The disorders in this group do not manifest with AME with a few exceptions. Fabry disease and CDG may present with strokelike episodes and seizures; Zellweger syndrome (peroxisomal defect) may present with a severe neurologic dysfunction in the newborn period. Group 1 disorders are not discussed in this chapter.
Group 2—intoxication: Inborn errors of intermediary metabolism lead to an acute and progressive intoxication due to accumulation of toxic metabolites proximal to biochemical defects. This group includes amino acid disorders (such as phenylketonuria (PKU), tyrosinemia, maple syrup urine disease [MSUD], homocystinuria), organic acidemias, urea cycle defects, galactosemia, and hereditary fructose intolerance. They present after a symptom-free interval either with acute symptoms such as vomiting, lethargy, acute encephalopathy, thromboembolic complications, liver failure, or chronic and progressive developmental delay. Some of these patients are at risk for recurrent metabolic crisis /encephalopathy.
Group 3—energy deficiency: These are the IEMs with symptoms due to a deficiency in energy production or utilization. In this group, there are glycogen storage disorders, gluconeogenesis defects, ketone utilization defects, fatty acid oxidation defects, primary lactic acidosis such as pyruvate dehydrogenase (PDH) or pyruvate carboxylase (PC) deficiency, as well as mitochondrial respiratory chain disorders. In energy deficiencies such as PDH, or PC deficiency there may be no symptom-free interval. The most frequent symptoms are generalized hypotonia, cardiomyopathy, and rapidly progressive neurologic deterioration.
This chapter focuses on group 2 and 3 disorders which can manifest with acute encephalopathy (Table 19.1).
■ INITIAL APPROACH TO DIAGNOSIS OF IEMs
Pediatric intensivists and neurologists are responsible for emergency care and stabilization of patients with IEMs and should be familiar with the fundamental aspects of such care and diagnostic approach.
Obtaining a careful past and current medical history including an assessment of diet, eating behaviors, and social, developmental, and family history is an essential first step (3). Historical features suggestive of IEMs include acute life-threatening symptoms in the newborn period, developmental delay, cardiomyopathy, episodic vomiting, liver dysfunction, hypoglycemia, acidosis, dehydration, and lethargy associated with minor illnesses. Diet history is crucial to determining food aversion. Patients with amino acid disorders or urea cycle defects may self-restrict their protein intake. A careful family history may reveal important clues that raise the possibility of IEM. Most IEMs are inherited as autosomal recessive traits, so there may be siblings with similar illnesses or deaths from sepsis with an unidentified pathogen or sudden infant death syndrome.
Routine biochemical tests such as basic metabolic panel and urine analysis can help to raise red flags for a possible IEM such as high anion gap acidosis or ketonuria. Once clinical suspicion is aroused based on the clinical presentation and basic laboratory investigation, more specific laboratory tests should be performed and supportive measures must be instituted immediately (4). The laboratory studies that should be obtained for a patient who has symptoms of a metabolic encephalopathy consistent with IEM are listed in Table 19.2.

How Does Newborn Metabolic Screening Help?
Since the early 1990s, tandem mass spectrometry has made screening possible for many IEM including amino acid, organic acid, and fatty acid oxidation disorders. The inclusion of these disorders in screening programs has enabled the timely diagnosis and treatment of these conditions and decreased complications such as mental retardation or death. Not all IEMs are screened in the newborn period. If the patient has symptoms and signs of a metabolic disorder, further testing should be performed even if the result of the newborn screening test was normal.
Which Biochemical and Clinical Findings Should Trigger Suspicion of IEMs?
IEMs are difficult to distinguish from one another since the same symptoms and signs occur in many different diseases. Symptoms and signs of the clinical presentation (neurologic deterioration, seizures, presence or absence of symptom-free interval after birth) in combination with biochemical tests help differentiate IEMs. Figure 19.1 provides a flowchart guiding IEM differentiation.
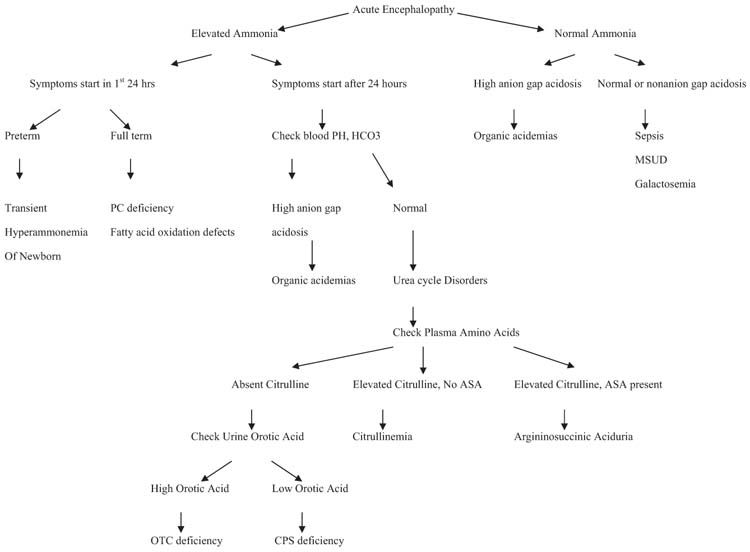
FIGURE 19.1 Diagnostic algorithm for acute metabolic encephalopathy.
While the symptoms and signs of each IEM vary considerably, I subdivide the most common disorders based mainly on their biochemical findings. Isolated persistent seizures causing encephalopathy do not have any biochemical findings in routine tests and they will be discussed under “seizures.” The clinician, prepared with such knowledge, can narrow the range of diagnostic possibilities, select the appropriate laboratory tests and treatment, and request a metabolic consultation.
■ INTOXICATION (GROUP 2) DISORDERS ARRANGED BY BIOCHEMICAL FINDINGS (A-D)
A—Metabolic Acidosis with High Anion Gap + Ketosis
Metabolic acidosis with high anion gap (> 16) is an important laboratory abnormality of many IEMs during acute episodes of illness. It is easily demonstrated by arterial blood gases or basic metabolic panel. Ketonuria is an important indicator of a metabolic disorder. Neonates do not normally produce much acetoacetate; ketosis detected in neonates by dipstick is always abnormal and should prompt serious consideration of an organic acidemia. Among the inborn errors, the largest group associated with high anion gap metabolic acidosis and ketosis is the group of organic acidemias such as propionic acidemia (PA) and, methylmalonic acidemia (MMA) (5,6).
Organic acidemias: MMA and PA are the most frequent forms of branched-chain organic acidurias, and caused by enzymatic defects in the catabolism of branched-chain amino acids (BCAAs) valine and isoleucine (Figure19.2) and of other propiogenic substrates, such as methionine, threonine, odd-chain fatty acids, and cholesterol.
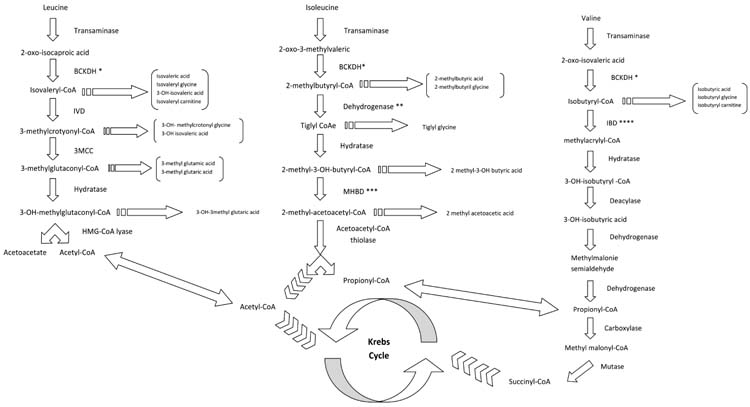
FIGURE 19.2 Pathways of branched-chain amino acids. *Branched-chain ketoacid dehydrogenase; **2-methylbutyryl Co-A dehydrogenase; ***2-methyl-30H-butyryl Co-A dehydrogenase; ****Isobutyryl-Co-A dehydrogenase.
PA is caused by a deficiency of propionyl CoA carboxylase (a biotin dependent enzyme). Isolated MMA results from either (1) deficient activity of the enzyme methylmalonyl-CoA mutase encoded by the MUT gene or (2) diminished synthesis of its cofactor adenosylcobalamine (cobalamin A or B disease). These autosomal recessive disorders can present with neurologic manifestations including acute progressive encephalopathy caused by the accumulation of toxic compounds proximal to the metabolic block. In most cases of PA and MMA, the onset of the disease is in the neonatal period but a later-onset presentation can occur.
Clinical and biochemical findings: The usual neonatal presentation is a sepsislike picture after an initial normal period of 24 to 48 hours: the neonate stops feeding well, begins vomiting, and becomes lethargic, which rapidly progresses to coma. Progressive neurologic findings include abnormal posturing and movements, hypotonia, and seizures, often with a burst-suppression electroencephalographic (EEG) pattern. If left undiagnosed and untreated, most of these patients die in the newborn period. Some patients, especially those with some residual enzyme activity, may survive this crash and go on to have other acute metabolic crises or develop chronic developmental and neurologic sequelae.
In addition to metabolic acidosis, and ketosis, these patients may also have elevated lactate caused by secondary interference with coenzyme A metabolism. Neutropenia, thrombocytopenia, and anemia can occur during acute crisis and may lead to false diagnosis of sepsis (6). Hyperammonemia is commonly but not uniformly seen in these patients.
In about one-third of patients, the disease presents with late onset after a symptom-free interval longer than 1 year. The clinical picture of late onset forms is variable, ranging from acute encephalopathy to intermittent or chronic symptoms such as neurodevelopmental delay, recurrent vomiting, and ataxia. The recurrent attacks are usually triggered by catabolic stress such as infections or increased intake of protein rich foods. Patients may be entirely normal between attacks. These symptoms are associated with metabolic acidosis, ketosis, and hyperammonemia as seen in the neonatal onset form (5).
Early diagnosis and treatment is crucial for a better outcome. Patients who are not diagnosed and treated effectively develop mental retardation, seizures, and acute and progressive dystonic syndrome that result from insult to basal ganglia.
Magnetic resonance spectroscopy may show a decrease N-acetylaspartate (NAA) (a sign of neuronal injury) and increase lactate. Magnetic resonance imaging (MRI) findings of PA and MMA are brain atrophy and abnormal myelination.
Urine organic acid analysis and plasma acylcarnitine profile are the first line diagnostic tests that identify abnormal metabolites (7). Plasma amino acid analysis shows nonspecific abnormalities such as hyperglycinemia and hyperalaninemia. In PA, urine organic acid analysis shows elevated levels of propionic acid, propionylglycine, tyglylglycine, 3-hydroxyl-propionate, and methylcitrate. Plasma acylcarnitine analysis shows elevated level of propionylcarnitine (7). Molecular genetic testing can be used to confirm the diagnosis. Propionyl CoA carboxylase enzyme activity can be measured in fibroblasts.
Patients with MMA show the same abnormal biochemical findings seen in PA patients in addition to elevated plasma and urine MMA level. Activity of methylmalonyl CoA mutase (Mut) can be assessed indirectly by measurement of incorporation of [1-14C] propionate into cellular macromolecules (C14 propionate tracer assay). Complementation analysis can determine the enzymatic block using heterokaryon rescue or enzymatic cross-correction (8).
B—Ketosis Without Acidosis or Hyperammonemia
Maple syrup urine disease: MSUD is caused by a deficiency of branched chain alpha ketoacid dehydrogenase (BCKD), which is the second enzyme in the degradation pathway of leucine, isoleucine, and valine (branched-chain amino acids [BCAAs]) (Figure 19.2). Accumulation of BCCAs, primarily leucine, is toxic and causes encephalopathy. Based on the clinical presentation, residual enzyme activity and thiamin responsiveness, 5 phenotypes have been described: (1) Classical MSUD is the most severe form and patients present with encephalopathy in the newborn period, (2) Intermediate MSUD with neurologic symptoms later in life but only rarely AME, (3) Intermittent MSUD with encephalopathy under catabolic stress intermittently, (4) Thiamine responsive MSUD, and (5) dihydrolipoamide dehydrogenase (E3) deficient MSUD with lactic acidosis and elevations of BCAAs and branched-chain keto-acid (BCKAs) (9). It is a combined deficiency of BCKD, PDH, and alpha ketoglutarate dehydrogenase complexes because E3 is a common component of these 3 enzymes (10).
Clinical and biochemical findings: Affected neonates with classical MSUD present with lethargy, poor suck, vomiting, and irritability after symptom-free interval of 2 to 3 days. If not diagnosed and treated, they develop opisthotonos, bicycling movements, and coma within 4 to 5 days. During acute encephalopathy, the EEG may demonstrate a V comb-like rhythm of 5 to 9 Hz spindle-like sharp waves over the central regions that are believed to be characteristic of MSUD.
Patients with MSUD do not have metabolic acidosis, hyperammonemia, or lactic acidosis (except E3 defects) but develop ketosis and ketonuria. Maple syrup odor is present in patients’ urine and cerumen. The presence of 2-ketoacids can be detected in urine with the 2, 4-dinitrophenylhydrazine (DNPH) test. Plasma amino acid analysis shows an elevation of the BCAA; the presence of alloisoleucine is a definitive marker of MSUD (10).
Patients with intermediate MSUD have very high levels of BCAAs and present with developmental delay and/or seizures in the first years of life. Episodes of AME are rare in this form.
Patients with intermittent MSUD have normal early development and have normal BCAAs when they are well. They present with episodic ataxia, lethargy, and acute encephalopathy triggered by stress such as fever, infection, surgery in the first years of life. During metabolic crisis, plasma amino acids and urine organic acid profile show elevated levels of BCAAs and BCKAs.
C—Acidosis, Ketosis, or Hypoglycemia Accompanied by Neurologic Deterioration
Glutaric aciduria type I: Glutaric aciduria (GA) type I is caused by a deficiency of glutaryl CoA dehydrogenase enzyme, which is involved in the catabolism of L-lysine, L-hydroxylysine, and tryptophan. Early clinical diagnosis and treatment is important to prevent irreversible brain damage (11).
Clinical and biochemical findings: Patients are usually asymptomatic in the first year of life. Macrocepahly is a common finding. Soft neurologic signs such as jitteriness and irritability may be present. The most common presentation is acute encephalopathy precipitated by an intercurrent illness such as fever, diarrhea, or other stresses such as immunizations or surgery in a relatively healthy 1 to 2 year of child. Patients develop sudden onset of hypotonia, loss of head control, seizures, opisthotonos, fisting, rigidity, and dystonia. The sequela of the acute encephalopathic crisis is bilateral neuronal degeneration of the caudate and putamen, and subsequently, a movement disorder. Patients are at risk for recurrent metabolic crisis during fever or respiratory or gastrointestinal illnesses (11).
During acute metabolic crisis patients can have acidosis, ketosis, hypoglycemia, hyperammonemia, and mild liver disease (Reye-like syndrome). Plasma amino acid profile is normal. Plasma acylcarnitine analysis shows elevated glutarylcarnitine (C5-DC). Urine organic acid analysis shows elevated levels of glutaric acid and 3-hydroxyglutaric acid (12).
MRI shows frontotemporal atrophy, collections of fluid over frontal lobe and at the base of brain, and arachnoid or subdural cysts. The fluid collection between the brain and cranium stretches bridging vessels, making them vulnerable to a minor trauma, so subdural hematomas may occur (11).
It is very important to prevent catabolism during intercurrent illnesses because it is the only way to reduce the frequency of acute encephalopathic crisis and irreversible striatal injury. Emergency treatment follows the basic treatment principles of metabolic diseases of the “intoxication” type, which is discussed below.
D—Hyperammonemia
The breakdown of proteins and other nitrogen-containing molecules results in the production of free ammonia. The urea cycle (Figure 19.3) detoxifies ammonia by converting it to urea. Urea cycle disorders are the most common cause of hyperammonemia, but other IEMs, especially organic acidemias, can cause a comparable derangement (Table 19.3). The pathogenesis for hyperammonemia in some of these conditions is not fully understood.
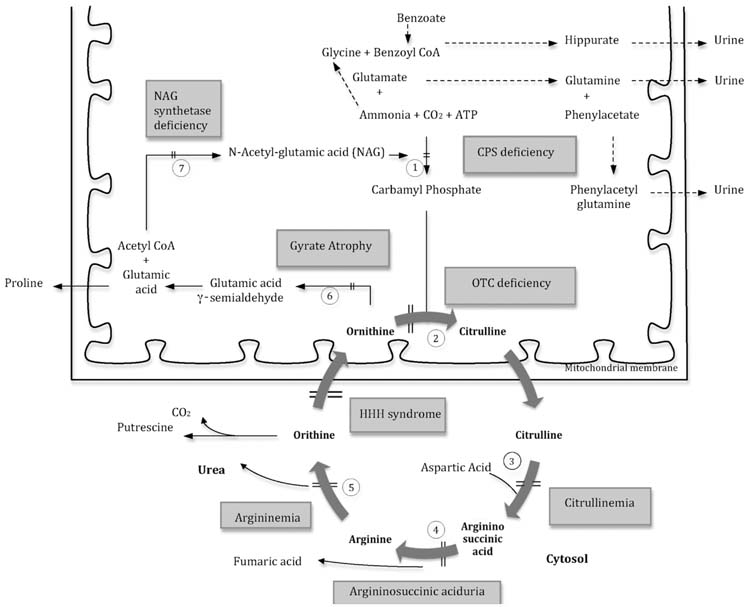
FIGURE 19.3 The urea cycle
Reactions shown with interrupted arrows are the alternate pathways for the disposal of ammonia. Enzymes: (1) carbamyl phosphate synthetase (CPS), (2) ornithine transcarbamylase (OTC), (3) argininosuccinic acid synthetase (AS), (4) argininosuccinic acid lyase (AL), (5) arginase, (6) ornithine 5-aminotransferase, (7) N-acetylglutamate (NAG) synthetase. Abbreviation: HHH syndrome, hyperammonemia-hyperornithinemia-homocitrullinemia syndrome.

Urea cycle disorders: The urea cycle includes 5 enzymes: (1) carbamyl phosphate synthetase (CPS), (2) ornithine transcarbamylase (OTC), (3) argininosuccinate synthetase (AS), (4) argininosuccinate lyase (AL), and (5) arginase. In addition to these enzymes, N-acetylglutamate (NAG) synthetase is needed to produce NAG, a cofactor of the CPS enzyme (13).
Clinical and biochemical findings: The clinical symptoms may appear in the neonatal period, or they may appear any time thereafter with varying degrees of severity. In the neonatal onset form, the affected neonate is normal at birth but presents with a sepsis-like picture by 24 to 48 hours of life. The baby stops feeding, begins vomiting, and displays lethargy that quickly progresses to coma. Physical examination may reveal hepatomegaly in addition to the neurologic signs of coma. The encephalopathy is characterized by increased intracranial pressure with a bulging fontanel and dilated pupils, and seizures are common (14).
The late-onset form may present in infancy, childhood, and adulthood with vomiting and neurologic abnormalities such as ataxia, mental confusion, agitation, irritability, and combativeness. These manifestations usually occur during a high protein intake or catabolic stress such as infections.
Routine laboratory tests often are normal. The blood urea is usually very low. Hyperammonemia may stimulate the brain stem respiratory center, leading to respiratory alkalosis. There may be mild increases in serum transaminases (alanine aminotransferase (ALT), aspartate aminotransferase (AST)), since ammonia can cause swelling of hepatic mitochondria (14).
It is imperative to measure plasma ammonia, amino acids, acylcarnitine levels, and urine organic acid levels in any ill infant whose clinical manifestations cannot be explained by sepsis. Brain ultrasound or computed tomographic imaging may show edema.
In infants with organic acidemias, hyperammonemia is commonly associated with high anion gap acidosis and ketonuria as discussed above.
The measurement of ammonia can be difficult with false elevations noted frequently. Ammonia should be measured in a free-flow blood sample and transferred to a laboratory on ice for immediate analysis to avoid artificial production from blood cells. In children and adults, the plasma ammonia concentration is usually less than 35 µmol/L. Blood concentrations in healthy newborns often can be as high as 100 µmol/L in a full-term baby and 150 µmol/L in a premature infant. The plasma ammonia concentration in the ill infant is usually more than 200 µmol/L.
A diagnostic algorithm for hyperammonemia is presented in Figure 19.1. The age of onset of symptoms, and presence or absence of acidosis help physicians to arrive at a diagnosis of hyperammonemia. Plasma amino acid analysis can help the diagnosis of different urea cycle disorders. Patients with deficiencies of CPS, OTC, or NAG synthetase have elevations in plasma glutamine and alanine with concurrent decrements in citrulline and arginine (14). A marked increase in urinary orotic acid in patients with OTC deficiency differentiates this defect from CPS deficiency. Differentiation in CPS vs. N-acetylglutamate (NAG) synthetase may require an assay of the respective enzymes, or mutation analysis. Clinical improvement occurring after oral administration of carbamylglutamate, however, may suggest NAG synthetase deficiency. Patients with deficiencies of AS, AL, or arginase have marked increases in the plasma levels of citrulline, argininosuccinic acid, or arginine, respectively (14).
General Aspects of Treatment of “Intoxication Type” Metabolic Disorders
The following principles of management are for all the disorders discussed above (group 2, subgroups A–D).
Management of acute metabolic decompensation: When IEMs are suspected, immediate treatment should be initiated even if the diagnosis is not yet established. AME, either at the onset or during a relapse of metabolic decompensation, is an emergency and patients should be managed in the intensive care unit. Delay in starting the emergency treatment results in rapid progression to encephalopathic crisis and irreversible brain damage. Serious neurologic damage is dependent on the duration of hyperammonemia and is very likely in infants with severe elevations in blood ammonia (> 300 µmol/L) for more than 12 hours.
There are 2 main goals of the acute treatment: (1) prevent catabolism by providing an adequate caloric intake and (2) remove toxic metabolites such as ammonia, organic acids, or leucine as quickly as possible.
After the first suspicion of an organic acidemia, urea cycle defect, or MSUD, protein intake should be discontinued and intravenous (IV) fluids containing glucose (glucose infusion rate at 10–15 mg/kg/min), electrolytes, and lipid (1–3 g/kg/day) should be started. It is essential to provide at least 1.5 times normal energy and fluid intake based on the patient’s age. It is important to minimize exposure to hypotonic fluids and maintain sodium levels of 138 to 145 mEq/L. Insulin can be added to keep the blood glucose levels below 150 mg/dL. Whenever possible, protein-free formula with high calories can be started via nasogastric or gastrostomy tube. The total base deficit should be calculated and corrected aggressively using IV bicarbonate solutions. In MSUD, parenteral nutrition consisting of BCAA-free L-amino acid mixture can be used in combination with glucose, lipid, electrolyte, and vitamins. The plasma levels of isoleucine and valine drop much faster than that of leucine and it is important to supplement isoleucine and valine (20–120 mg/kg/day) after 1 day of MSUD treatment. Maintaining the normal levels of isoleucine and valine facilitates the normalization of plasma levels of leucine (9).
In critically ill patients with severe acidosis, hyperammonemia, or high leucine level who are unresponsive to treatment within the first few hours, hemodialysis should be initiated. All neonatal patients with severe neonatal hyperammonemic coma (plasma levels greater than 600 µmol/L) should be hemodialyzed. Continuous venovenous hemofiltration is more effective in ammonia, BCAAs/BCKAs, or organic acid removal than peritoneal dialysis.
Scavenger therapy offers an important way to decrease ammonia in a form other than urea. Compounds such as sodium benzoate and phenylacetate (250–500 mg/kg/day) lower blood ammonia by forming complexes with glycine and glutamine, respectively (13). Both are available in oral and IV forms.
Arginine is a nonessential amino acid in healthy individuals, who form it in the urea cycle, but it is essential in most patients with urea cycle disorders (except arginase deficiency). For this reason, patients with urea cycle defects are likely to need a supplementation of arginine (210–660 mg/kg/day) to maintain the levels within normal range (50–200 µmol/L). Patients with OTC deficiency benefit from supplementation with citrulline (170 mg/kg/day). Administration of arginine or citrulline is contraindicated in patients with arginase deficiency (13).
Sodium benzoate, sodium phenylacetate, and arginine can be administered together for maximal therapeutic effect. A priming dose of these compounds is followed by continuous infusion until recovery from the acute hyperammonemic crisis (Table 19.4). The recommended therapeutic doses of both compounds deliver a substantial amount of sodium to the patient that should be calculated as part of the daily sodium requirement. A commercial, parenteral preparation of sodium benzoate plus sodium phenylacetate is available (Ammonul). Sodium benzoate and sodium phenylbutyrate can also be used if hyperammonemia is persistent in organic acidemias. Recently, carbamylglutamate (50–100 mg/kg/day) has been shown to improve hyperammonemia in PA patients (15).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


