
■ OVERVIEW OF ACUTE DEMYELINATING SYNDROMES
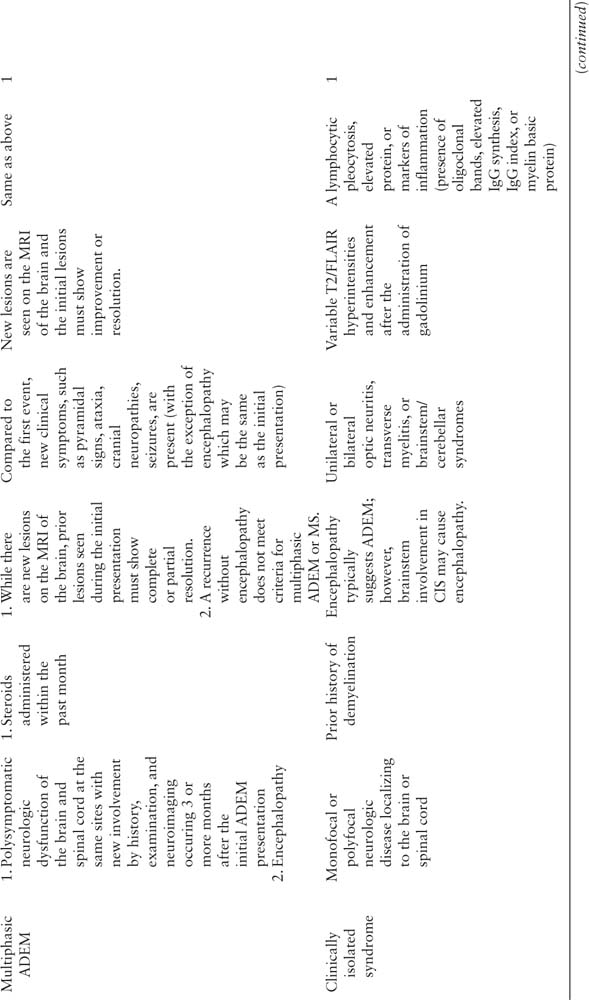
Acquired demyelinating syndromes may be isolated or recurrent events. If a child is presenting with their first neurologic event, the diagnosis is either ADEM or a clinically isolated syndrome. If the child is encephalopathic and polysymptomatic, he or she meets criteria for ADEM (Figure 14.1) (1). Otherwise, an individual’s first episode is referred to as a clinically isolated syndrome. A clinically isolated syndrome must not involve encephalopathy, but may involve either monofocal symptoms and signs (related to dysfunction at one CNS location) or multifocal symptoms and signs (localizing to multiple CNS areas) (1). A clinically isolated syndrome may present as optic neuritis, transverse myelitis, or brainstem, cerebellar, or hemispheric dysfunction.
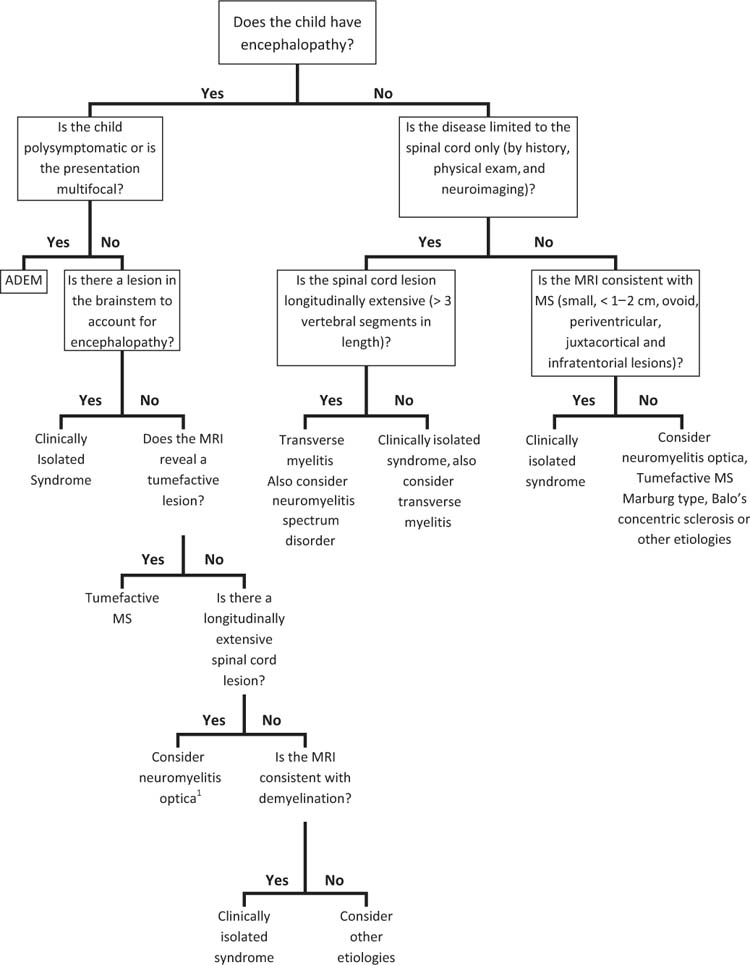
FIGURE 14.1 Flowchart to guide the diagnosis of the first demyelinating event in a child.
1 While many children present with the classic findings of visual dysfunction or spinal cord symptoms, neuromyelitis optica may present with encephalopathy, seizures, or cranial nerve findings.
If a child has a history of a demyelinating event, the diagnosis may be MS, NMO, recurrent ADEM, multiphasic ADEM, recurrent optic neuritis, recurrent transverse myelitis, or chronic relapsing inflammatory optic neuropathy (CRION). These diagnoses will be reviewed later in this chapter.
All demyelinating diseases are diagnosed by clinical signs and symptoms. However, often laboratory tests and neuroimaging are used to further define the extent of the disease. It is not always possible during an acute episode to confirm the diagnosis or establish the prognosis, which may not be apparent until follow-up testing is performed or there is a recurrence.
ADEM, NMO, transverse myelitis, and fulminant demyelinating diseases (acute hemorrhagic leukoencephalitis, Marburg variant, Balo concentric sclerosis, and tumefactive MS) are more likely to present to the intensive care unit (ICU). MS flares rarely require ICU admission, and isolated optic neuritis flares do not require ICU treatment; however, these are common demyelinating diseases and will be covered as well.
■ ACUTE DISSEMINATED ENCEPHALOMYELITIS
Introduction
ADEM has been considered the prototypic demyelinating disease in children as most first-time demyelinating events in children were labeled as ADEM. It is typically a monophasic disease, but relapses and even the subsequent development of MS have been reported. In an effort to better classify those with ADEM versus a suspected first presentation of MS (called a clinically isolated syndrome), consensus definitions were proposed by the International Pediatric Multiple Sclerosis Study Group in 2007 (1). The working definition of ADEM, not yet validated, requires a polysymptomatic presentation and encephalopathy. It is considered inaccurate to call all first demyelinating events ADEM if the event does not meet these two criteria.
Demographics
While ADEM can occur at any age, it is more common in children; especially prepubertal children (mean age 5–8 years). Males and females are equally affected, perhaps with a slight male predominance (2). There does not seem to be a racial or ethnic predominance.
Clinical Features
Children often have a prodrome of generalized malaise/fatigue, fever, headache, nausea, and vomiting prior to the onset of neurologic symptoms (3–6). Encephalopathy, manifested as fatigue or irritability to frank coma, is required for the diagnosis of ADEM according to the International Pediatric Multiple Sclerosis Study Group (1). Furthermore, the definition requires a polysymptomatic and multifocal presentation. Symptoms depend on the sites of inflammation; however, in general, motor symptoms are frequent, with 60% to 95% of children having unilateral or bilateral pyramidal signs (4). Ataxia, cranial neuropathies (including optic neuritis), and seizures are also common. If optic neuritis is present, there is often bilateral involvement. Sensory symptoms are less frequently reported. This may be due to pathophysiologic differences between ADEM and other demyelinating diseases (see Multiple Sclerosis, below) or simply because the severity of other neurologic symptoms and encephalopathy compromise the sensory examination.
Classically, ADEM has been diagnosed following an antecedent illness or vaccination in the month prior to the onset of neurologic symptoms. However, such an immune trigger is not always recognized, and a history of infection or vaccination is not required for the diagnosis of ADEM (2,4).
Imaging
A head computed tomography (CT) reveals multiple lesions in the white matter, but typically does not capture the extent of disease and may even be normal (5,7,8). The appearance of lesions on MRI is variable in ADEM. Most children present with diffuse fluid-attenuated inversion recovery (FLAIR) or T2-weighted hyperintense lesions of brain and spinal cord that are large (> 1–2 cm) and asymmetric with poorly defined margins. Basal ganglia and thalamic involvement may occur, with or without other supratentorial or infratentorial lesions. Spinal cord involvement is also common. Smaller lesions (< 5 mm) resembling MS are also possible.
Enhancement is variable. Some have proposed that all lesions should enhance indicating their acute nature since this would demonstrate a monophasic time course. In contrast, in MS there may be both enhancing and nonenhancing lesions suggesting prior episodes and a more chronic recurrent process. However, in some patients with ADEM both enhancing and nonenhancing lesions may occur, so the presence of some nonenhancing lesions should not alter the diagnosis. In addition, the development of lesions on MRI may lag behind the clinical symptoms in ADEM. Similarly, the MRI may reveal new or enlarging lesions within the first 3 months, even in the setting of clinical improvement.
Cerebrospinal Fluid
A lymphocytic pleocytosis (mean 51 cells/mm3, range 0–270) or elevated protein (mean 73.9 mg/dL, range 45–120) was seen in 70% to 75% of patients who underwent a lumbar puncture (2,5,7). Other markers of CNS inflammation, such as the presence of oligoclonal bands, immunoglobulin G (IgG) synthesis, or myelin basic protein may be present in the cerebrospinal fluid (CSF), especially during the acute phase. According to some series, oligoclonal bands are present in 4% to 29% of children with ADEM (2,3,5,7), but they were not present in any patient with ADEM in another large study (9).
Prognosis, Recurrent ADEM, and Multiphasic ADEM
The prognosis is generally favorable, and most children with ADEM make a full recovery without sequelae. Residual deficits depend on the clinical sites involved, and the most common deficits are motor problems, visual impairment, cognitive impairment, behavioral problems, and seizures (4,7).
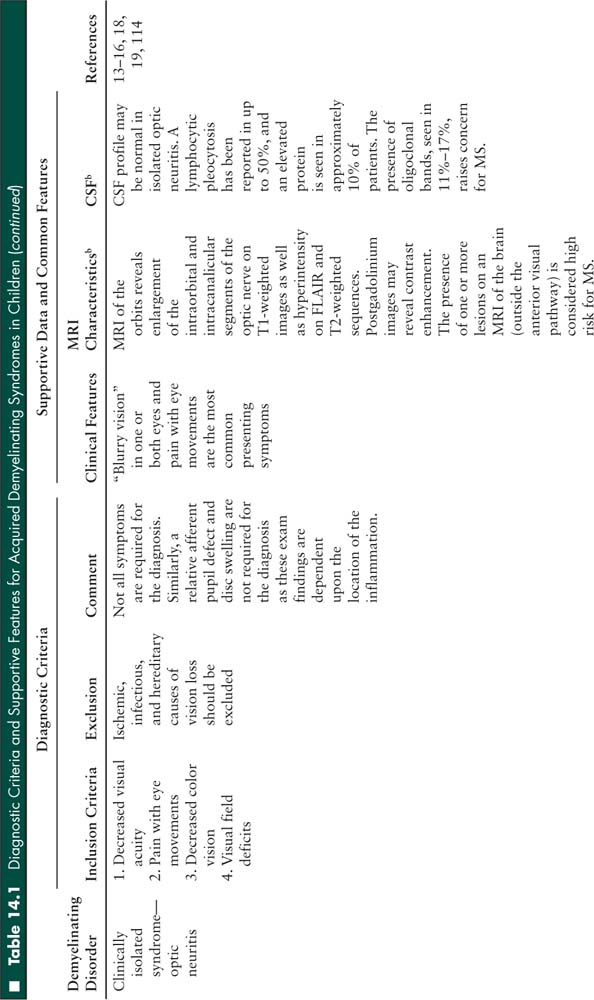
Relapse may occur after a first neurologic event consistent with ADEM. If symptoms recur within the first 3 months, this is considered part of the initial presentation and does not qualify as a relapse (Figure 14.2). Similarly, symptom recurrence within 1 month of discontinuing steroids is also considered part of the initial event, and is not considered a relapse. In contrast, neurologic symptoms occurring 3 months after the first episode and more than 1 month after discontinuing steroids may be due to recurrent ADEM, multiphasic ADEM, or MS. If the neurologic symptoms are exactly the same as the initial event (although there may be expansion of an existing lesion), the diagnosis is recurrent ADEM. If new symptoms or MRI lesions appear, the diagnosis is multiphasic ADEM. For both recurrent and multiphasic ADEM, the presentation must include encephalopathy. If encephalopathy is not present, a clinically isolated syndrome is the likely diagnosis at the time of the relapse. MS requires 2 non-ADEM events; therefore, a single relapse after ADEM that does not fulfill criteria for ADEM currently does not meet MS criteria (1).
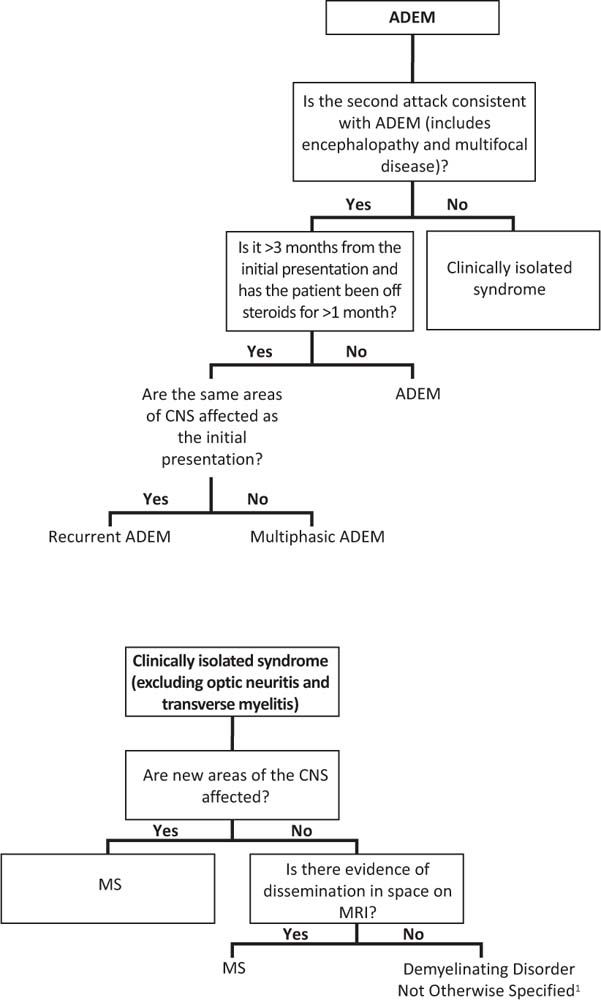
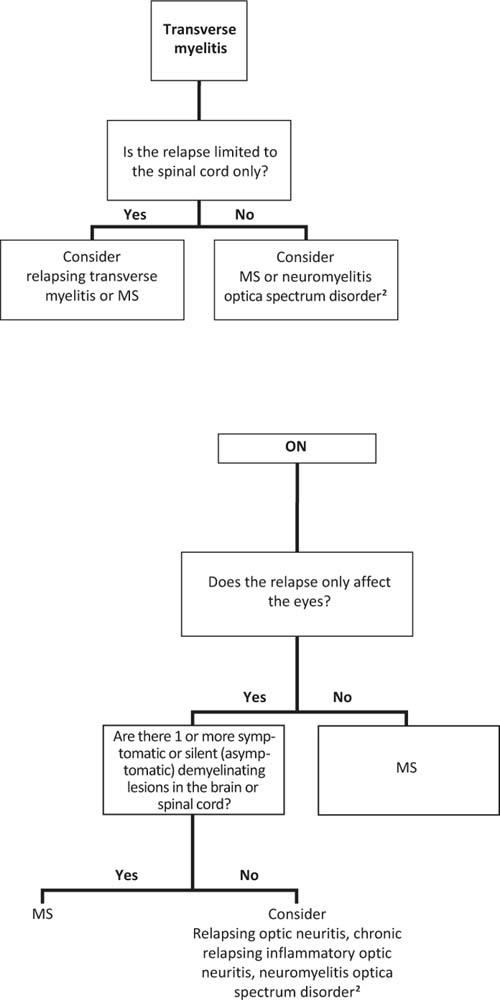
FIGURE 14.2 Flowchart to guide the diagnosis of recurrent demyelinating diseases. The pathways depend on the initial diagnosis.
1 A relapse of a clinically isolated syndrome affecting the same area of the central nervous system fulfills dissemination in time; however, dissemination in space is required for a definition of pediatric multiple sclerosis.
2 The diagnosis of neuromyelitis optica (NMO) requires the presence of both optic neuritis and spinal cord disease. In children who have one but not both criteria, especially in the setting of relapsing disease that is not consistent with multiple sclerosis, NMO immunoglobulin G (IgG) should be sent. If the serum NMO-IgG is positive the diagnosis is NMO spectrum disorder.
■ ACUTE HEMORRHAGIC LEUKOENCEPHALITIS
Introduction
Acute hemorrhagic leukoencephalitis or acute hemorrhagic encephalomyelitis (AHL or AHEM) is thought to be a fulminant form of ADEM. It may be distinguishable from typical ADEM on head CT or MRI by the presence of edema, mass effect, and hemorrhages (8).
Clinical Features
Similar to ADEM, AHL may follow a respiratory infection in some patients (2); however, a prior illness is not seen in all patients and is not required for the diagnosis. A prodrome of fever, headache, or vomiting may be present for 1 to 2 days prior to the onset of neurologic symptoms (10,11). Altered mental status is present, and various neurologic symptoms, such as weakness and seizures, occur depending on the location of the lesions. Pupillary asymmetry and vital sign instability occurs in the setting of herniation syndromes.
Imaging
Often, the lesions are large and confluent, and they may be unilateral or bilateral (8). A head CT may reveal cortical edema with midline shift or cerebellar involvement with brainstem compression and obstructive hydrocephalus. MRI of the brain may reveal hemorrhages using FLAIR or gradient echo techniques (8), although small, petechial hemorrhages may not appear on MRI.
Cerebrospinal Fluid
A granulocytic pleocytosis is seen in the CSF, and erythrocytes may be present (8). Compared to ADEM, the CSF white blood cell (WBC) and protein counts are often higher in AHL (median CSF WBC 145 and protein 115) compared to ADEM (median CSF WBC 15 and protein 32) (10). Elevations in the CSF IgG synthesis and myelin basic protein, or presence of oligoclonal bands, are also possible. The opening pressure is often elevated.
Prognosis
For AHL, death within 1 week has been reported (2) and 60% of pediatric cases are fatal (10). However, survival may be possible with aggressive treatment (10,11). Two surviving cases with pathologic confirmation were treated with decompressive craniotomies after failed medical management of elevated intracranial pressure (ICP) (10,11). Corticosteroids and other therapies, such as intravenous immunoglobulin (IVIg), have also been used.
■ CLINICALLY ISOLATED SYNDROMES
Introduction
A clinically isolated syndrome is a first demyelinating event in a child or adult that does not meet criteria for ADEM. This is an important distinction for 2 reasons. First, clinically isolated syndromes have a higher likelihood than ADEM of converting to MS. Second, disease modifying therapy is recommended for children and adults presenting with a clinically isolated syndrome who are “high-risk” for the conversion to MS since it delays the onset of MS; therefore, early recognition is essential.
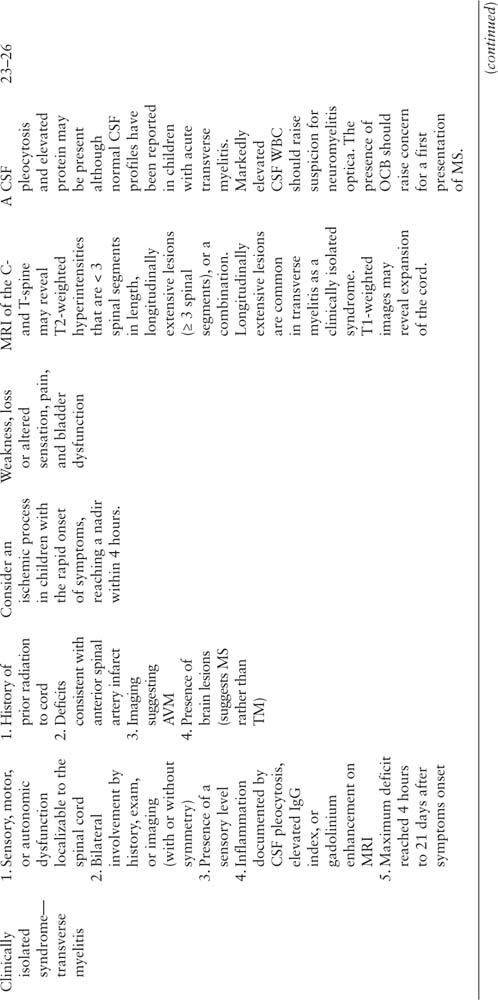
Clinically isolated syndromes are a heterogeneous group of disorders, defined by the affected area within the CNS. The clinical presentation may be monofocal or multifocal. However, in contrast to ADEM, encephalopathy is not present (1), with one exception. If the brainstem is affected, encephalopathy may be present due involvement of the reticular activating syndrome. In this example, the patient may still meet criteria for a clinically isolated syndrome. The two most common clinically isolated syndromes are optic neuritis and transverse myelitis (12).
Optic Neuritis
Introduction
Optic neuritis is a common acquired demyelinating syndrome in children affecting the anterior visual pathway (optic nerve and chiasm). Optic neuritis can be an isolated clinical event without other neurologic signs and symptoms or associated with disseminated demyelination (as in ADEM, MS, or NMO). If isolated optic neuritis is present, the child would not be admitted to the ICU. However, optic neuritis could occur in the context of additional symptoms, and visual impairment may not be appreciated if encephalopathy is present. Optic neuritis may be unilateral or bilateral, and the distribution is defined by clinical history, physical examination, and neuroimaging.
Demographics
Bilateral optic neuritis is more common in younger children. Excluding children with ADEM, 72% of children < 10 years of age present with bilateral optic neuritis whereas 70% of children ≥ 10 years present with unilateral optic neuritis (13).
Clinical Features
The clinical presentation includes decreased visual acuity, contrast sensitivity, and color vision; visual field defects; and pain with extraocular movements in one or both eyes. There is considerable variation in the clinical features. For example, some children may have normal or slightly decreased visual acuity while others have no light perception (14,15). While subtle clinical features are possible, approximately two-thirds of children present with visual acuities of 20/200 or worse (16). The physical exam may reveal a relative afferent pupillary defect in unilateral cases. Fundoscopic exam may reveal disc edema when the inflammation occurs at the papilla (papillitis); however, swelling may not be present in retrobulbar optic neuritis and is therefore not required for a diagnosis. A formal neuroophthalmologic examination is often helpful in confirming the diagnosis and extent of inflammation.
Imaging
MRI of the orbits reveals enlargement of the intraorbital and intracanalicular segments of the optic nerve on T1-weighted images as well as hyperintensity on FLAIR and T2-weighted sequences. Postgadolinium images may reveal contrast enhancement. An MRI of the brain is also indicated, even in the absence of other neurologic signs or symptoms, as the presence of additional FLAIR and T2 hyperintensities outside the optic nerves and chiasm significantly increases the likelihood of developing further clinical attacks and a diagnosis of MS (see Prognosis and the section on Multiple Sclerosis, below) (13). In the United States, 54% of neurologists surveyed about practice patterns also obtain imaging of the cervical and thoracic spine (17). Similar to silent brain lesions, the presence of asymptomatic cord lesions would raise the possibility for MS or NMO, especially in cases of bilateral optic neuritis with longitudinally extensive cord lesions (see below), although such lesions are typically symptomatic.
Cerebrospinal Fluid
A lumbar puncture is not always performed in cases of isolated optic neuritis. In a study assessing practice patterns of U.S. neurologists, more than two-thirds of those surveyed routinely order CSF studies, such as oligoclonal bands or IgG index (17). A lymphocytic pleocytosis has been reported in about 43% to 50% of patients (15,18,19), and elevated protein has been reported in about 10%. In general, oligoclonal bands are seen in about 11% to 17%. The lumbar puncture is perhaps more helpful in patients with optic neuritis and normal MRI of the brain (excluding the optic nerves) as adults with oligoclonal bands in the CSF had an increased likelihood of developing MS (20).
Prognosis
The visual prognosis for children with optic neuritis is excellent. Despite often severe inflammation, most patients recover to a visual acuity of 20/40 or better with a short-course of intravenous (IV) steroids (14) (see treatment, below), although some recover without intervention. Optic neuritis may be a clinically isolated syndrome and never recur or may be the presenting symptom of MS or NMO. MRI scans (brain, C-, and T-spine) and laboratory studies (NMO IgG) can be helpful in identifying patients at risk for disease progression. Optic neuritis can also recur without brain or spine involvement, called sequential or recurrent optic neuritis depending on the affected eye and timing of relapse (in relation to the initial presentation and administration of an oral steroid taper).
CRION has been reported in adults and children (21,22). Initially described in adults, the optic neuritis in CRION is bilateral sequential (both eyes affected within days to weeks of each other) although relapses may occur years later in either eye. Clinically, there is severe pain and poor visual acuity, and MRI scans of the brain are typically normal, all of which help differentiate adult optic neuritis associated with CRION from optic neuritis typical of MS (21).
Transverse Myelitis
Introduction
Transverse myelitis is a demyelinating disease affecting the spinal cord. It is characterized by acute or subacute motor, sensory, and/or autonomic spinal cord dysfunction. Often, spinal cord inflammation occurs in the context of another demyelinating disease, such as ADEM, MS, or NMO, but may also occur as a clinically isolated syndrome. This section will cover transverse myelitis as a clinically isolated syndrome, and the differences between transverse myelitis as a clinically isolated syndrome and the other demyelinating diseases with spinal cord involvement will be highlighted. The terms complete and incomplete transverse myelitis have been used inconsistently; therefore, it is perhaps preferable to avoid such terms and rather describe the clinical and radiographic features.
Demographics
One study of 47 children with acute transverse myelitis at an academic center reported a bimodal incidence in toddlers (< 3 years of age, representing 38% of patients with transverse myelitis), and teenagers (> 11 years, 49%) (23). The remaining 13% were between ages 5 and 8 years. In this study, males and females were equally affected, and no correlation was seen between presentation and time of year. At another pediatric academic center where 38 patients were evaluated, only 5% of children were under the age of 3 years (24). In contrast to the prior study, more females were affected, especially over the age of 10 years. Similarly, when the cases were restricted to only transverse myelitis as a clinically isolated event (ie, without brain lesions or subsequent diagnosis of MS), more females were affected (all ages). In this study, 66% of children presented in colder months (November–April). The differences in demographics between these pediatric studies may be due to varying methodologies and inclusion criteria or referral practices for each academic institution.
Clinical Features
The presence of motor deficits, pain, bladder dysfunction, and sensory involvement should prompt consideration of acute transverse myelitis. A thoracic sensory level is often detected on examination, meaning sensory changes are seen below an identifiable level. However, all spinal levels (cervical through sacral) have been reported (23). Symptoms often progress over days (range 4 hours to 21 days). Symptom nadir typically occurs within 10 days for most patients (25). More rapid progression of symptoms (nadir in less than 4 hours) should raise concern for an ischemic process (26).
Acute transverse myelitis may be a clinically isolated syndrome in which only spinal cord symptoms and signs are present, or it may occur in the setting of MS (spinal cord and brain symptoms) or NMO (spinal cord and optic neuritis).
Imaging
On MRI axial and sagittal T2-weighted images the cord may reveal a single lesion or multiple lesions (< 3 spinal segments in length), longitudinally extensive lesions (≥ 3 spinal segments), or a combination (23,24). Longitudinally extensive lesions are common in ADEM, transverse myelitis (as a clinically isolated syndrome), and NMO, whereas smaller, asymmetric lesions with discrete borders are more common in MS (24). T1-weighted images may reveal expansion of the cord. While the clinical presentation often includes a thoracic sensory level, the MRI burden of disease is often more expansive than suggested by the clinical symptoms (23). Thus, a patient with a thoracic sensory level could have a lesion extending from the cervical cord to the conus. Normal MRI scans of the spinal cord have been reported in children with transverse myelitis (23,24). In such cases, localizable symptoms to the cord and inflammatory CSF profiles are used to establish the diagnosis of transverse myelitis (24).
MRI of the brain and orbits are helpful in assessing the extent of demyelination, especially in patients without referable brain symptomatology (ie, encephalopathy, cranial nerve deficits, seizures, etc), to determine risk for MS and NMO.
Cerebrospinal Fluid
A CSF pleocytosis and elevated protein may be present but are not required for the diagnosis. Normal CSF profiles have been reported in children with acute transverse myelitis (23,24). Markedly elevated CSF WBC should raise suspicion for NMO. The presence of oligoclonal bands is rare in acute transverse myelitis as a clinically isolated syndrome and may indicate that transverse myelitis is the first presentation of MS or a component of ADEM, and an MRI of the brain, if not previously performed, is warranted.
Prognosis
The recovery from neurologic deficits is influenced by associated demyelinating syndromes. Recovery is better if the spinal cord symptoms and signs occur in the setting of ADEM. In MS, spinal cord involvement may improve with time but could also result in persistent symptomatology, such as paresthesias. One pediatric study compared recovery when spinal cord demyelination occurred in the context of MS or as a clinically isolated syndrome. All MS patients were fully ambulatory with milder scores using the Expanded Disability Status Scale. Prognosis was not as favorable in acute transverse myelitis as a clinically isolated syndrome. Roughly 33% of patients recover without sequelae or minor neurologic symptoms, 33% have moderate deficits, and 33% have severe deficits. When part of a clinically isolated syndrome, children with motor and sensory deficits as well as bladder dysfunction were more likely to have permanent paralysis compared to children with partial syndromes (motor and/or sensory and/or bladder, but not all 3 symptoms) (24). In addition to motor symptoms, bladder dysfunction is a common long-term complication from acute transverse myelitis.
In adults with acute transverse myelitis, poor recovery was associated with rapid symptom progression, back pain, and spinal shock (26–30); however, rapid progression was not associated with poor outcome in a pediatric study (23). The same pediatric study reported that prompt diagnosis of acute transverse myelitis (< 7 days) resulted in improved outcomes (23); however, time to treatment was not a factor in another pediatric study (24).
There have been a few pediatric deaths in children with acute transverse myelitis. High cervical cord lesions may result in respiratory failure (23). As many children are critically ill due to flaccid paralysis and respiratory difficulties, the acute treatment may be complicated by the failure of other systems. One death was reported during the acute hospitalization due to complications from a pulmonary embolus (24).
Approximately 70% to 90% of cases of acute transverse myelitis are truly isolated events that do not relapse or progress (25). There are a number of factors that help differentiate monophasic transverse myelitis from a first presentation of MS. The presence of one or more of the following is suggestive of a first presentation of MS: (1) predominantly sensory symptoms with motor preservation, (2) asymmetric examination, (3) discrete spinal cord lesions (less than two spinal segments), (4) positive oligoclonal bands in the CSF, and (5) an abnormal MRI of the brain (26). Longitudinally extensive MRI lesions, especially in the presence of optic neuritis or deep gray/brainstem lesions, should prompt consideration of NMO. Finally, transverse myelitis can relapse without brain MRI lesions or other features suggestive of MS or NMO (31).
■ MULTIPLE SCLEROSIS
Introduction
MS is the prototypical chronic demyelinating disease, and is one of the most common causes of neurologic disability in the world. Classically, MS has been diagnosed clinically by evidence of two CNS lesions “typical of MS” disseminated in time and space (32). More recently, criteria have been refined to include evidence from MRI (33,34). Acute, potentially devastating variants of MS include Marburg type, tumefactive MS, and to a lesser degree, Balo concentric sclerosis.
Demographics
In prepubertal children, MS affects males and females equally, or perhaps has a slight male predominance (35). However, postpuberty, there is a 2:1 female to male ratio. A higher proportion of African American and Hispanic children have been reported in pediatric MS compared to adult MS (36–38).
Clinical Features
The clinical symptoms of pediatric MS are quite variable depending on the areas of the brain and spinal cord affected. Pediatric MS most commonly presents with sensory symptoms (39,40) including numbness, tingling, or burning, present for more than 24 hours (and typically lasting 2–4 weeks), or optic neuritis (36). Such cases would not be admitted to the critical care unit. Other clinically isolated syndromes (such as some cases of transverse myelitis, as detailed above, or brainstem lesions) are more likely to require critical care observation and treatment. Children may also have motor problems, cranial neuropathies, cerebellar signs, vestibular disease, bowel and bladder involvement, cognitive problems, or fatigue.
Imaging
MS lesions classically appear hyperintense on T2-weighted or FLAIR imaging and hypointense on T1-weighted imaging. MS lesions are commonly found in the periventricular white matter, corpus callosum, junctions of the gray-white matter (called juxtacortical), and the spinal cord (41–44). The presence of one or more periventricular lesions is predictive of MS (45). Discrete lesions (with well-defined borders) and lesions perpendicular to the corpus callosum are also highly suggestive of pediatric MS (46) but may not be present on an initial MRI (45). Brainstem lesions (47,48) and large tumorlike lesions exhibiting mass effect (49,50) have been reported in fulminant cases (see below).
In general, the anatomic location and size of T2-weighted hyperintense lesions correlate well with histopathologic findings (51). Over time, the hypointensity of T1 lesions may increase or decrease, based on the permanence or reversibility of the process (52). Chronically, T1-hypointensity in MS lesions (called “black holes”) are associated with severe white matter destruction, including axonal loss (53). T1-lesion load correlates well with changes in neurologic disability (54). Moreover, in children, the presence of T1 lesions, especially such lesions that are present on repeat imaging, are highly predictive of MS (45).
In adults, new lesions in MS typically enhance for an average of 3 weeks (range 1–13 weeks) (55). However, contrast-enhancement of lesions is not necessarily captured at the time of a first attack. At the time of the initial presentation, only 25% to 70% of children had enhancing lesions (45,46).
Cerebrospinal Fluid
A lymphocytic pleocytosis (median 8/µL, range 0–61/µL) is seen in about two-thirds of children with MS (56). Protein may also be elevated. The presence of oligoclonal bands is estimated between 40% to 90% of children with pediatric MS (57).
Prognosis
Recovery from MS flares is variable. Without treatment, symptoms may resolve within a few weeks. The use of corticosteroids may hasten recovery (58). While randomized controlled trials investigating the use of corticosteroids in adults with MS flares have confirmed their benefit in the treatment of acute symptoms, their long-term effect on disease progression or disability has not been proven (59,60).
Compared to adults, children experience more relapses, especially within the first few years of disease onset (38). This finding was confirmed when adjusted for sex, race, and disease modifying therapy. A natural history study revealed a median time to secondary progressive MS of 28.1 years in children versus 18.8 years in adults (61). While pediatric MS is more slowly progressive compared to adult-onset MS, children reach disability endpoints at earlier ages (41.4 years of age versus 52.1 years in adults). Frequent relapses and disability at younger ages were also seen in earlier studies (40,62).
Fulminant Disease—Marburg Type
Introduction
Marburg type of MS was originally described in 1906 as an MS variant with a fulminant course (63,64). The original cases were described as showing “hypercellular demyelinating plaques, with oedema, faint astroglial reaction, and presence of hypertrophic and giant astrocytes” (65). This fulminant demyelinating disease presents with acute lesions appearing to be of the same age in the context of a rapidly, progressive clinical course. Pathologically, there is extensive macrophage infiltration (47). Despite case reports detailing the pathology, imaging, and biochemical features of aggressive cases of demyelinating disease, Marburg type remains best defined clinically by its rapidly progressive development and evolution of neurologic deficits. There has been one case of “acute multiple sclerosis” in a child, although this was likely Balo concentric sclerosis (see below) (66).
Clinical Presentation
Marburg type may present similarly to exacerbations or relapses of more typical forms of MS. Initial neurologic signs and symptoms may be focal or multifocal with variable severity. The rapid worsening of neurologic signs and the emergence of new deficits over the course of days to weeks is a hallmark of Marburg type. Neurologic signs may include gross motor and sensory disturbances, involvement of the anterior visual pathway, bulbar dysfunction, and areflexia (47). Respiratory failure may necessitate mechanical ventilation (67).
Imaging
Head CT may be normal (68) or may show a large hypodensity in the white matter or multiple hypodensities (63). Brain MRI reveals single or multiple hypointensities on T1-weighted images and hyperintensities on T2-weighted or FLAIR images, often with rim or diffuse enhancement (65,69).
Cerebrospinal Fluid
Similar to other demyelinating diseases, a pleocytosis, elevated protein, or markers of inflammation (the presence of oligoclonal bands, or elevated IgG synthesis or myelin basic protein) may be present.
Prognosis
Many patients die within a few months despite aggressive therapy (63,68,70). While survival has been reported, such cases may not truly reflect Marburg type (69,71).
Fulminant Disease—Balo Concentric Sclerosis
Introduction
Balo concentric sclerosis has long been considered a rare and acute variant of MS. In 1928, Balo originally described the condition in a 23 year-old law student presenting with aphasia and absent cremasteric reflexes, followed by diffuse neurologic signs, including facial weakness, hemiplegia, and incontinence, resulting in death (72,73). Autopsy revealed concentric lesions with alternating layers of normal and degenerated white matter, leading Balo to call the entity “encephalitis periaxialis concentric” (72).
Clinical Features
Many patients present with the acute onset of weakness, although a variety of neurologic symptoms, such as sensory symptoms, visual deficits, cognitive problems, and bulbar dysfunction, has been reported (74). The clinical manifestations are dependent on the regions of the brain affected.
Imaging
The MRI of the brain reveals a concentric pattern of hypodensities and hyperdensities in the white matter. After contrast administration, a thin border of enhancement is seen at the periphery of the lesion (8), and this is the key to differentiating Balo concentric sclerosis from other demyelinating disorders.
Cerebrospinal Fluid
Similar to other demyelinating diseases, a pleocytosis, elevated protein, or markers of inflammation (the presence of oligoclonal bands, or elevated IgG synthesis or myelin basic protein) may be present.
Prognosis
Early in the characterization of the disease, many adult cases were found to have resulted in death within weeks to months following onset. However, before the advent of MRI, when definitive diagnosis relied on postmortem pathologic study, patient selection for biopsy was based on the severity of the disease, allowing for the possibility of missing cases of Balo concentric sclerosis with a more benign course, thereby precluding diagnosis. Correlation of modern neuroimaging studies with pathologic findings has enabled antemortem diagnosis of Balo concentric sclerosis. This has lead to a trend in recent reports of Balo concentric sclerosis cases exhibiting prolonged survival, spontaneous remission, and an overall more benign disease course (74,75).
Pediatric Cases
Balo concentric sclerosis has been described in a 13- year-old female, who presented with hemianopsia and anomia (76). She had an elevated CSF WBC count (30 cell/µL) and large lesions with alternating hypointensities and hyperintensities on MRI in a concentric pattern with enhancement. She was also found to be positive for human herpesvirus 6. She was initially treated with IV methylprednisolone and foscarnet. Six months later, she deteriorated and was retreated with the same medications; however, she continued to progress and IVIg was initiated. She received IVIg monthly with clinical improvement and no further relapses.
There is one other reported case of a child with acute multiple sclerosis, published in 1965 in the case records of the Massachusetts General Hospital in the New England Journal of Medicine (66). A 6-year-old female presented 1 week after a brief respiratory illness and 3 days after a fall from a “horizontal bar” without loss of consciousness. Neurologic symptoms began hours after the fall, including blurry vision, and progressed over the next few days to include left hemiparesis, slurred speech, a right gaze preference, and she became unresponsive. Her temperature rose to 108F. Her CSF did not reveal cells or an elevated protein, although the initial lumbar puncture revealed “a pressure of 100 mm.” She was unresponsive to pain with fixed, unresponsive pupils, arm extension with increased tone, and flaccid paralysis of the lower extremities. For the increased pressure, neurosurgical intervention was required, and she had bilateral burr holes placed. She was treated with methylprednisolone, antibiotics, and phenytoin. She had multisystem organ failure requiring respiratory, cardiac, nutritional, and hematologic support. She developed seizures and further increases in ICP. Due to the poor prognosis, she was extubated. She died on hospital day 31. Pathology revealed diffuse perivascular lesions throughout the brain that varied in size and had clearly demarcated areas of demyelination. In addition, some of the “smaller lesions had a striking concentric pattern suggestive of altering zones of greater and lesser intensity of myelin.” A dense cellular infiltrate consisting of macrophages was seen in larger lesions. She was diagnosed with acute multiple sclerosis.
Fulminant Disease—Tumefactive MS
Introduction
MS typically presents with small, discrete lesions in the periventricular, juxtacortical, or infratentorial brain. However, other patients have larger lesions in the brain with surrounding edema on head CT or brain MRI. Such lesions are called tumefactive and the imaging features may raise concern for a malignancy, often resulting in a biopsy. However, clinical and radiographic characteristics suggestive of demyelination are described below to help distinguish a demyelinating disorder from a malignancy.
Clinical Features
Symptoms depend on the location of the tumefactive lesion. For example, acute weakness, cranial nerve involvement (including optic neuritis), and ataxia may be seen. Patients may present with headaches, seizures, nausea, and vomiting, which are uncommon in MS.
Imaging
Head CT may be normal or reveal hypodensities of the white matter. Patchy rim enhancement may be present on a postcontrast head CT. On T2-weighted or FLAIR MRI, lesions appear hyperintense in the periventricular white matter in older children. The lesions in younger children are present in the subcortical white matter. Rim enhancement of the lesion may be complete or incomplete. Greater enhancement may be seen on the side facing the ventricle (77). Spinal cord lesions may also be present.
Cerebrospinal Fluid
There are few reports of CSF findings in tumefactive MS in children. An elevated CSF WBC was seen (range 12–63 cells/µL), all had a normal CSF protein (77,78). In a single study, oligoclonal bands were present in one of the three children (78).
Prognosis
For some children, the disease is monophasic, without relapses, and the term “tumefactive MS” is a misnomer. However, for other children, tumefactive demyelination occurs during the course of MS. In other words, the initial presentation may be typical of MS; however, a relapse may include a tumefactive lesion.
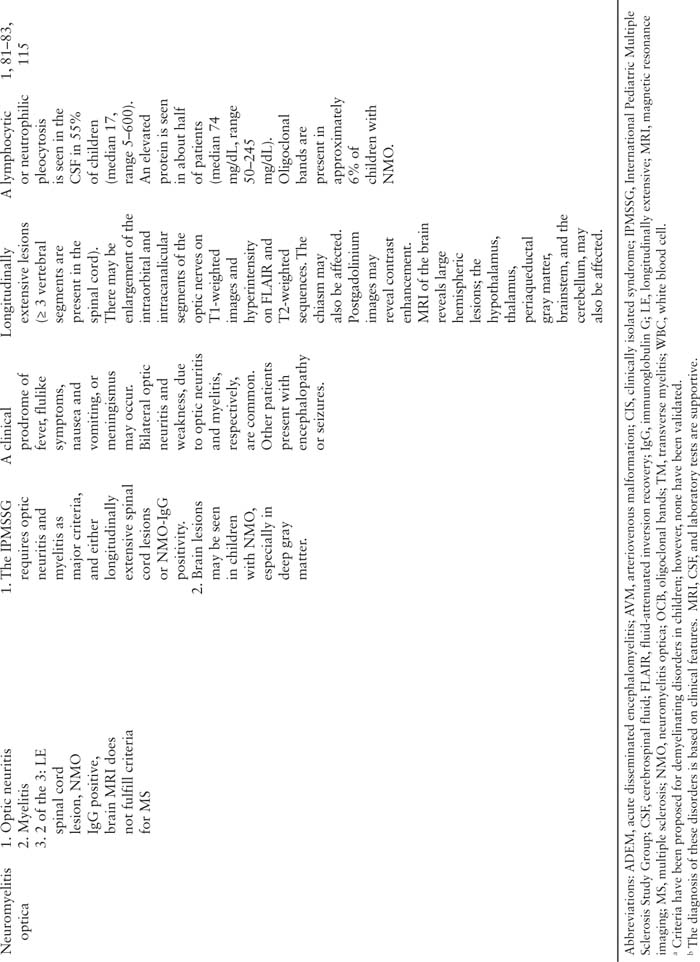
■ NEUROMYELITIS OPTICA
Introduction
NMO (also known as Devic disease) is a demyelinating disease with predilection for the optic nerves and spinal cord. Initially thought to be a variant of MS, NMO is now believed to be a different demyelinating disease, largely due to the identification of a serum biomarker, IgG autoantibody (NMO-IgG), which may be present in the CSF as well (79,80). The clinical features of optic neuritis and spinal cord involvement are required for the diagnosis, which may be confirmed by the presence of the NMO-IgG antibody. Children who do not meet the clinical criteria, but are NMO-IgG positive, are often diagnosed with “NMO spectrum disorder.”
Demographics
NMO spans the pediatric age spectrum, from 2–18 years (81). Females are predominantly affected (81,82). Two pediatric studies have noted a higher proportion of African-American children (34%–66%) compared to white children (27%–33%). Hispanic children are also affected (10%–33%) as well as Native American children (9%) (81,82). While NMO was previously thought to be more common in Asian adults, there were no Asian children in these cohorts.
Clinical Features
A prodrome of fever, viral illness, nausea, vomiting, hiccups, or meningeal signs may precede neurologic symptoms (81). Visual disturbance and weakness from optic neuritis and spinal cord disease, respectively, are the most common presenting symptoms (82). Vision loss is often bilateral and severe. Symptoms localizable to the brain, such encephalopathy, seizures, cranial nerve findings (other than optic neuritis), are common and should not preclude a diagnosis of NMO.
Diagnosis
While the evaluation is similar for most acquired demyelinating syndromes (see below), there are particularly important laboratory studies. First, there is an IgG autoantibody (NMO-IgG) that has been identified in the serum and CSF of pediatric and adult patients with NMO (79). The NMO-IgG antibody binds to an aquaporin-4 channel in the astrocytic foot processes in the blood-brain barrier. The presence of antibody in the serum (which is preferred) or CSF is not necessary for the diagnosis, which requires the presence of transverse myelitis and optic neuritis. Therefore, those with a negative NMO-IgG antibody may still be diagnosed with NMO (or NMO spectrum disorder if diagnostic criteria are not met). NMO-IgG positivity is dependent upon treatment status, and may even appear later in the disease after an initial negative test (82).
NMO is associated with many other autoimmune diseases, such autoimmune rheumatologic, thyroid, and gastrointestinal diseases, and elevated antibodies (antinuclear antibodies and antiphospholipid antibodies) (83). Therefore, additional laboratory studies are required to further investigate these conditions.
Imaging
Spine MRI typically reveals longitudinally extensive lesions (≥ 3 vertebral segments); however, similar lesions are seen in ADEM, transverse myelitis, and, less frequently, MS (84). As described above, the optic nerves may reveal enlargement of the intraorbital and intracanalicular segments on T1-weighted images and hyperintensity on FLAIR and T2-weighted sequences. Postgadolinium images may reveal contrast enhancement. Children with NMO also have involvement of the optic chiasm. Brain MRI may reveal large hemispheric lesions or smaller lessions in the hypothalamus, thalamus, periaqueductal gray matter, brainstem, or cerebellum.
Cerebrospinal Fluid
A pleocytosis is seen in the CSF in 55% of children (median 17, range 5–600, with 50% lymphocytic and 50% neutrophilic) (81). While many patients have a low CSF WBC, others will have a markedly elevated CSF WBC, which should raise suspicion for NMO. Protein elevations are also seen in about half of patients (median 74 mg/dL, range 50–245 mg/dL). Oligoclonal bands are present in approximately 6% of children with NMO (81).
Prognosis
NMO is a relapsing disease, and recovery after an attack is often incomplete. Many children have residual weakness or visual impairment. NMO is associated with higher disability than MS in children. Death has been reported in adults with NMO (5-year survival of 68%) due to respiratory failure in the setting of brainstem disease (85,86). Fortunately, pediatric deaths due to NMO have not been reported.
Unique to NMO, there are comorbid or associated diagnoses that warrant further investigation. The hypothalamic-pituitary axis may be affected resulting in menstrual irregularities and the syndrome of inappropriate antidiuretic hormone. In addition, as noted above, patients with NMO often have symptoms or serologic evidence of other autoimmune diseases.
■ GENERAL APPROACH TO SUSPECTED DEMYELINATING DISEASE
History
The history of patients with demyelinating disease is markedly heterogeneous, even within the specific clinical entities. Nevertheless, a detailed history is critical to prompt disease recognition and the initiation of treatment. Isolated or multifocal neurologic signs and symptoms localizing to the CNS should give rise to increased clinical suspicion for these conditions. Symptoms may involve motor, sensory, cranial nerve, brainstem, cerebellar, cognitive, and/or autonomic deficits. Motor symptoms may localize to cerebral white matter, brainstem, or spinal cord. Deficits may result in weakness or paresis and may be focal or multifocal. Sensory deficits may include numbness or abnormal pain, temperature, or vibratory sensation. Lhermitte sign, which is an “electric-shock–like” sensation running down the spine or limbs associated with neck flexion or extension, may be present and is pathognomonic for cervical spinal cord disease. Bowel, bladder, and sexual dysfunction may be present in various forms and degrees of severity.
Seizures are more common in ADEM than MS. Isolated sensory symptoms, which are unlikely to require ICU care, are often present in MS whereas pyramidal involvement is more likely in ADEM, transverse myelitis, and NMO. Optic neuritis may be seen in ADEM, MS, NMO, infectious diseases, and other inflammatory or autoimmune disorders, or present as a clinically isolated syndrome.
Following a presentation suspicious for acute demyelination, the subsequent history should attempt to further support or refute specific etiologies and explore other potential diagnoses (Table 14.2). A history of prior demyelinating events is supportive of MS or NMO, or rarely, recurrent or multiphasic ADEM. Recent viral infection or vaccination is more commonly reported in ADEM and transverse myelitis than MS, although the exacerbation of MS symptoms can occur with an increase in body temperature (Uhthoff phenomenon). Such episodes may occur with a fever or illness or occur with exercise, hot showers, baths, or saunas, or hot weather.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


