Anaemia
Anaemia is defined as having a haemoglobin (Hb) less than the lower limit of the reference range for age (Table 29.1).
Clinical features suggestive of anaemia
- Pallor.
- Poor growth.
- Pale conjunctivae.
- Signs of cardiac failure.
- Flow murmur.
- Listlessness.
- Lethargy.
- Shortness of breath.
Investigation
If anaemia is suspected, begin with a full blood examination (FBE), blood f lm, ferritin and reticulocyte count. The initial classification is based on the mean corpuscular volume (MCV) (see Fig. 29.1).
Iron deficiency
Iron deficiency is common among Australian children, but it is often subclinical. Anaemia only occurs in those with more severe deficiency. Iron deficiency may be present in 10–30% of children in high-risk groups. Iron deficiency (see Table 29.1) may lead to impaired cognitive and psychomotor performance, even in the absence of anaemia.
Most cases of iron-deficiency anaemia in young children are due to inadequate dietary intake, although faecal blood loss may be provoked by cow’s milk protein. In adolescent girls, blood is lost through menstruation (Table 29.2).
In iron-deficiency anaemia, the ferritin, Hb, mean corpuscular volume (MCV) and mean corpuscular Hb concentration (MCHC) are low. In mild iron deficiency, the FBE is normal but the ferritin is reduced, which demonstrates low iron stores. Ferritin is an acutephase reactant and may be misleadingly normal/high during an acute febrile illness. In such circumstances, a repeat measurement a month later or an empiric trial of iron therapy are alternative strategies.
Iron studies are frequently requested, but in otherwise well, community-based children, results other than ferritin contribute little to the diagnosis. Serum iron concentration, total iron binding capacity (TIBC), serum transferrin and transferring saturation are not clinically useful. Serum iron concentration varies considerably throughout the day and week in normal individuals, is low in chronic disease and as an acute phase response.
Remember that iron-deficiency anaemia secondary to a poor diet may be associated with other macro-and micronutrient deficiencies.
Table 29.1 Haemoglobin reference ranges for age
| Age | Lower limit of normal range of Hb (g/L) |
| 2 months | 90 |
| 2–6 months | 95 |
| 6–24 months | 105 |
| 2–11 years | 115 |
| >12 years | girls – 120 boys – 130 |
Prevention of iron deficiency
- Introduce iron-containing solids from 4–6 months.
- Avoid cow’s milk in the first 12 months of life (apart from small amounts in custards and cereals).
- Cow’s milk should only form a small part of the diet up to 2 years of age.
- Ensure that formulas (if used) and cereals are iron fortified.
- Consider supplementation in high-risk groups (see Table 29.2).
Good sources of iron for children
See also chapter 6, Nutrition.
- Infant milk formulas.
- Fortified breakfast cereals.
- Meat (including red meat, chicken and fish).
- Green vegetables (especially legumes, e.g. peas and beans).
- Dried beans and fruit.
- Egg yolk.
Note: Iron absorption from non-meat sources is increased when taken with foods high in vitamin C (citrus fruit, strawberries, caulifl ower and broccoli).
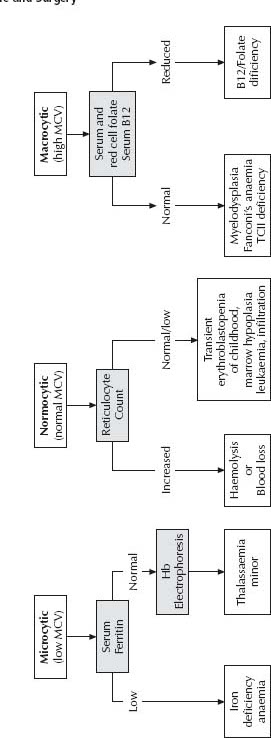
Table 29.2 Children at high risk for iron-deficiency anaemia
| Group | Additional risk factors | Mechanisms |
| <6 months of age | Prematurity | Inadequate stores |
| Low birth weight | ||
| Multiple births | ||
| Maternal iron deficiency | ||
| 6–24 months of age | Exclusive breast-feeding after 6 months | Inadequate intake |
| Delayed introduction of iron-containing solids | Cow’s milk may cause microscopic gut blood loss | |
| Excessive cow7#8217;s milk | ||
| Adolescents | Females | Menstruation |
| Poor diet | Rapid growth spurt | |
| Aborigines Migrant families Socially disadvantaged Vegetarian/fad diets | Poor diet | Inadequate intake |
| Excessive cow’s milk |
Management
- Dietary advice should be given in all cases. This includes increasing the amount of iron containing foods and limiting the intake of cow’s milk. Parents often need considerable support to manage behavioural issues around the diet, especially in the toddler age group.
- Supplemental iron should be recommended for any child with documented iron deficiency, even in the absence of reduced haemoglobin, because it takes a long time to replenish iron stores by dietary change alone. Behavioural changes and an improved feeling of well being are often noted with iron replacement even in children who were not anaemic.
- For young children, supplemental iron is usually given as ferrous gluconate mixture (daily dose 1 mL/kg of the 300 mg/5 mL preparation) and should be continued for 3 months after the Hb has returned to normal to replenish stores. Alternatives to ferrous gluconate mixture are shown in Table 29.3. The stools may become black/grey. As iron overdose can be fatal, supplements should be stored in a locked cabinet.
- Parenteral iron supplementation and blood transfusions are rarely indicated in children. Blood transfusions may be used if a very anaemic child requires urgent surgery or if cardiac failure is present. Transfusion should be slow and only raise the Hb to 60–80 g/L (see Calculating the blood transfusion volume, p. 372).
Vitamin B12 deficiency
B12 deficiency in childhood most commonly presents during the first 2 years of life. The most common cause is nutritional, due to undiagnosed maternal B12 deficiency in a fully breast-fed child. Transcobalamin II deficiency is uncommon, but is associated with normal serum B12 levels, despite severe tissue B12 deficiency. Rarer inherited metabolic causes of cellular B12 deficiency exist and diagnosis can be masked by therapy. Early involvement of a metabolic physician is imperative for timely diagnosis of these rare conditions.
Table 29.3 Alternatives to ferrous gluconate mixture

Any child with failure to thrive or neurodevelopmental abnormalities with an associated haematological abnormality (any cytopenia, macrocytosis or hypersegmented neutrophils) should be suspected of B12 deficiency and investigated urgently. The urgency relates to the propensity for rapid neurological deterioration (seizures, apnoea, choreoathetosis) and the lack of reversibility of these symptoms if treatment is delayed.
Investigations
- Bone marrow aspirate can confirm megaloblastosis within an hour to allow therapy to commence immediately.
- Serum homocysteine and urinary methylmalonic acid are also important to establish cellular vitamin B12 deficiency.
Haemoglobinopathies
β-Globin thalassemia
β–Thalassemia minor is common and causes hypochromic microcytosis without significant anaemia or clinical symptoms. One clue is an elevated red cell count on the FBE. The diagnosis is confirmed by Hb electrophoresis (or high performance liquid chromatography, HPLC) demonstrating an elevated HbA2. Iron deficiency obscures the diagnosis by reducing HbA2 levels, and hence children with thalassemia minor are often unnecessarily treated with iron therapy.
Thalassemia major is now uncommon because of the increased use of antenatal screening and prenatal termination. However, the diagnosis should be considered in the context of hypochromic microcytic anaemia presenting in the second 6 months of life (i.e. after â-globin chain switch has occurred at 6 months). Hepatosplenomegaly, marked erythroblastosis and bizarre red cell forms on the blood film are usually diagnostic.
α-Globin thalassemia
α-Thalassemia traits are relatively common in Asian populations. Hb Barts (four-gene deletion) classically presents with hydrops fetalis at birth. HbH disease (three-gene deletion) usually presents with mild to moderate microcytic anaemia but children are asymptomatic when well. When physiologically stressed, they can become significantly anaemic. Children with á-thalassemia traits (one-or two-gene deletions) may be microcytic (from birth) but not anaemic and are asymptomatic throughout life.
Sickle cell anaemia
Homozygous SS or double heterozygous (HbS/â–thalassemia trait) usually presents after 6 months of age (after â-globin chain switch).
Clinical presentations include:
- Anaemia (haemolysis or aplasia).
- Joint pains (especially small hand and foot joints).
- Acute chest syndrome (pneumonia-like with prominent hypoxia).
- Arterial ischaemic stroke.
- Acute splenic sequestration (rapidly progressive anaemia and splenic enlargement).
- Painful crisis (usually bone or abdominal pain).
- Asymptomatic diagnosis when parents are known carriers.
- Sepsis, especially encapsulated organisms in young infants.
Diagnosis
Diagnosis is made by blood film examination, sickle solubility tests and Hb electrophoresis.
Management
Long-term management includes folate supplementation, penicillin prophylaxis, appropriate vaccinations, and hydroxyurea or transfusion support as required. Acute management of crisis includes hydration, analgesia, transfusion and often antibiotics. Chest syndrome and stroke usually require exchange transfusion. Specialist consultation is required for each presentation.
Haemoglobin C/E
HbC and E are common in Asian populations, and both the heterozygous and homozygous forms are asymptomatic. Blood films may show many target cells. Thalassemia minor and HbC or E double heterozygotes present clinically as thalassemia major.
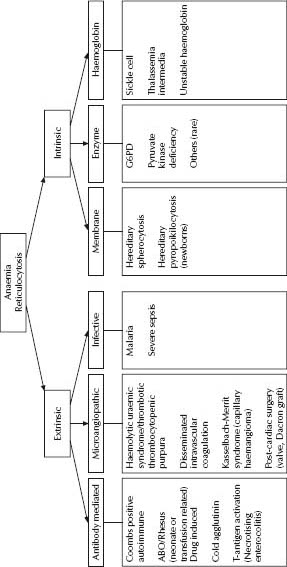
Haemolysis
Acute haemolysis in childhood is a life-threatening disorder that usually requires admission, thorough investigation and potentially transfusion support. Severe anaemia can develop quickly and frequent clinical review of vital signs and monitoring of Hb is required.
Diagnostic features of haemolysis include anaemia, polychromasia on the blood film, reticulocytosis and hyperbilirubinaemia. Haptoglobin is unhelpful in infants.
Investigations
First line investigations include:
- Blood film examination:
– Spherocytes: hereditary spherocytosis, Coombs positive, ABO, glucose-6-phosphate dehydrogenase (G6PD).
– Fragments: microangiopathic haemolysis.
– Blister/bite cells: oxidative haemolysis (drug or G6PD).
– Sickle cells.
- Direct Coombs test.
- Heinz body preparation.
- Hb electrophoresis/isopropanol test for unstable Hb.
- G6PD assay.
- Eosin 5 maleimid (E 5M) screening test for hereditary spherocytosis.
Further investigation is often required once the acute episode has resolved, and usually requires input from a specialist.
Potential causes of acute haemolysis are shown in Fig. 29.2.
Transient erythroblastopenia of childhood
This is a form of acquired red cell aplasia predominantly affecting children <7 years old.
Anaemia is normochromic, normocytic and there is no reticulocyte response until the recovery phase commences. Bone marrow aspirate may show red cell aplasia with preservation of other cell lines. The prognosis for previously normal children is excellent, with recovery for most within 2 months. No specific therapy is warranted and blood transfusion is best avoided if possible (consider if Hb <50 g/L and no reticulocyte response). The differential diagnosis is Blackfan–Diamond syndrome, which usually occurs at a younger age, but may be indistinguishable on peripheral blood and bone marrow aspirate findings.
Severe bleeding disorders in childhood can present at any time, although it is usually in the newborn period. Spontaneous bleeding or bruising of multiple ages in unusual sites should raise suspicions of a bleeding tendency. Often coagulopathy screening is required to differentiate a bleeding diathesis from non-accidental injury. Family history, drug history and history of previous surgical challenges (including tooth extraction) are important. Routine coagulation screening preoperatively in well children is rarely indicated and coagulation testing should be guided by the clinical history.
Investigation
First line investigations of a suspected bleeding disorder include:
- Platelet count and blood film.
- Activated partial thromboplastin time (APTT).
- Prothrombin time (PT) or international normalised ratio (INR).
- Fibrinogen.
Interpretation of these investigations is shown in Table 29.4. Further investigations should usually be done in discussion with a haematologist.
Fibrinogen is an acute-phase reactant. In severe sepsis, when fibrinogen should be elevated, a normal level is still consistent with disseminated intravascular coagulation (DIC).
If the investigations above are normal in the setting of clinically abnormal bleeding, consider factor XIII deficiency, Von Willebrand disease, platelet function defects or a capillary fragility syndrome. Specific investigations will be required.
Table 29.4 Investigation of coagulation abnormalities
| Screening test result | Causes |
| Low platelet count | Idiopathic (or immune) thrombocytopenic purpura (ITP) |
| Neonatal alloimmune thrombocytopenia (NAITP) | |
| Congenital thrombocytopenia syndromes | |
| Chemotherapy/marrow replacement | |
| Isolated prolonged APTT | Factor XI, IX, VIII deficiency |
| Von Willebrand disease | |
| Heparin | |
| Factor XII (no clinical bleeding) | |
| Isolated prolonged PT/INR | Factor VII deficiency |
| Warfarin | |
| Prolonged APTT, PT | Liver disease |
| Low fbrinogen | Disseminated intravascular coagulation (DIC) ± also low platelets Vitamin K deficiency, factor II, V, X deficiency (normal fibrinogen) |
In the setting of acute bleeding, if the diagnosis of a specific bleeding disorder is unclear, give 10–20 mL/kg of fresh frozen plasma (FFP) ± platelets.
Generally, children presenting acutely with bleeding disorders should be discussed with a haematologist. Haemophilia A or B, von Willebrand disease and other bleeding disorders are complex disorders requiring specialist management.
General measures
These are applicable to all congenital bleeding disorders.
- Analgesia:
– Do not give aspirin or other NSAIDs.
– Narcotic analgesics should only be given as part of a comprehensive pain management plan to avoid overuse.
- Do not give intramuscular injections. Do not do arterial puncture.
- Lumbar punctures should only be done after haematological consultation and appropriate factor replacement.
- Splinting limbs reduces pain.
- Consult with a haematologist about the need for joint aspiration. Beware of the risk of:
– Volkmann’s ischaemic contracture in forearm bleeds.
– Femoral nerve palsies with retroperitoneal bleeds tracking underneath the inguinal ligament.
Haemophilia A (factor VIII deficiency)
Management of bleeding
- Dosage of factor VIII is usually 30–50 units/kg, which increases factor VIII levels by 60– 100%. Note that 1 unit/kg of factor VIII raises levels by 2%. Repeat doses are usually required 8–12 hourly.
- Most bleeding can be controlled with a single dose calculated to increase the factor VIII level to 50%.
Note: A minor head injury can become serious: the factor VIII level should be raised to 100% and the child admitted for observation.
- Recombinant human factor VIII is readily available for haemophilia treatment centres and used for all patients.
- Patients with factor VIII inhibitors may be treated with recombinant factor VIIa (Novoseven). The usual dose of factor VIIa is 90–100 mcg repeated in 2 h.
Mouth bleeding
Use tranexamic acid tablets (see Pharmacopoeia).
Haemophilia B (Christmas disease, factor IX deficiency)
Bleeding is treated with factor IX concentrate (Monofix).
- Requirements: as in haemophilia A.
- Dose: in general 1 unit factor IX/kg increases levels by 1.6%.
- Frequency: injections at 24 h intervals (the half-life for factor IX is 24 h).
Von Willebrand disease
Responds to cryoprecipitate, or to human-derived factor VIII (Biostate). Maintaining adequate supply of Biostate in Australia is difficult and therefore its use usually requires specialist haematologist intervention. Many patients respond to desmopressin (see pharmacopoeia). The half-life of von Willebrand factor is approximately 4 h, but factor VIII levels continue to be increased for 48–72 h after the infusion of cryoprecipitate. Further doses are given if bleeding recurs.
Idiopathic thrombocytopenic purpura
Idiopathic thrombocytopenic purpura (ITP), also known as immune thrombocytopenic purpura, is an acquired thrombocytopenia due to shortened platelet survival (immune mediated) in the absence of other disturbances of haemostasis or coagulation.
In young children, ITP usually presents with bruising and petechiae, often with a history of recent viral infection. In some instances there is oral bleeding, epistaxis, rectal bleeding or haematuria.
Bone marrow biopsy is not necessary where the clinical presentation, FBE and blood film suggests ITP (i.e. no hepatosplenomegaly or lymphadenopathy; no anaemia, leucopenia or blasts).
Although bruising and petechiae can present dramatically, morbidity in ITP is usually minimal. Intracranial haemorrhage is the most serious risk but the probable incidence is <1%.
Management
Controversy surrounds the indications and best form of treatment for children with acute ITP. Without active treatment, most patients’ platelet counts return to a satisfactory level within a month.
Careful observation without specific treatment may be appropriate in milder cases. Patients with active bleeding (e.g. mucosal and gastrointestinal) should receive treatment to increase their platelet count more rapidly. Some authorities also recommend treating patients with a platelet count <10–20 × 109/L and a florid petechial rash (especially mucosal petechiae).
When treatment is indicated, corticosteroids are usually first line therapy. High dose i.v. immunoglobulin (IVIG) is reserved for the most severe or refractory cases. Various steroid regimens have been used. The following has been demonstrated to raise the count almost as quickly as IVIG: prednisolone 4 mg/kg for 1 week (maximum 75 mg/day), then 2 mg/kg for 1 week followed by 1 mg/kg for 1 week.
While the platelet count is very low, the child should rest quietly at home. As the count rises, more activity is allowed, but contact sports, cycling and rough physical activity should be avoided until the count is normal. Strictly avoid aspirin, NSAIDs and intramuscular/subcutaneous injections (including immunisations) until thrombocytopenia remits.
It is common for thrombocytopenia to recur with further viral infections in the year after diagnosis. Chronic ITP (lasting >6 months) occurs in <10% and requires specialist management. Treatment options include splenectomy and the use of rituximab (monoclonal antiCD20 antibody). The risk–benefit ratio should be carefully considered and discussed with a haematologist.
Anticoagulation therapy in children
Thromboembolic disease is increasingly frequent in neonates and children, presumably because of increased survival of children with previously fatal primary disorders and the increased use of invasive arterial and central venous catheters, as well as extracorporeal circuits. Consequently the frequency of anticoagulant use in children is increasing, for both treatment and primary prophylaxis.
The coagulation systems of children and adults are physiologically different, which impacts on the use of investigations and the action of anticoagulant medications. Current literature suggests children receiving anticoagulant therapy appear to have higher treatment failure rates and more bleeding complications than adults. Thus anticoagulation in children should be managed only by specialist paediatric haematologists.
The most commonly used anticoagulant agents in children are unfractionated heparin (UFH), low-molecular-weight heparin (LMWH) and warfarin.
- UFH is given i.v. and is advantageous because of its rapid onset of action, short half-life and reversibility. However, there is controversy over appropriate therapeutic ranges in children and the interpretation of currently available monitoring tests. Heparin-induced thrombocytopenia appears to be significantly less in children than in adults.
- LMWH is frequently used because of its more predictable bioavailability and weight-adjusted dosing schedules, and reduced need for therapeutic monitoring. A number of LMWH preparations are available, but most experience in children is with enoxaparin (Clexane). Twice daily subcutaneous dosing is recommended, but the inability to completely reverse LMWH once given, limits its use in very sick children. Extreme care must be taken when children are having procedures (e.g. lumbar puncture), and at least two doses should be omitted before any procedure. APTT does not reflect the anticoagulant activity of LMWH and may be normal in a fully anticoagulated child. The generally accepted therapeutic range is 0.5–1.0 anti-factor Xa units. Like UFH, LMWH is excreted via the kidneys and half-life may be prolonged in renal disease.
- Warfarin is the agent of choice for long-term therapy, but there is a multitude of paediatric-specific issues. There is no paediatric preparation, resulting in complicated dosing schedules. Avoidance of half or quarter tablets can be achieved by variable daily doses (e.g. alternating daily doses, 1 mg one day and 2 mg the next). Monitoring is now significantly easier with ‘point of care’ capillary blood monitors, although the accuracy is operator dependent and technical limitations remain (especially at higher therapeutic ranges). Lifestyle restrictions are necessary for children on long-term warfarin (mostly avoidance of contact sports), which may impact on adherence.
- Thrombolytic therapy in children is reported to cause major bleeding (intracranial or requiring transfusion) in >10%, with variable treatment success rates reported. Discuss this with a paediatric haematologist.
Blood transfusion is common in the tertiary paediatric setting. The most common blood product transfused is packed red cells or packed cell concentrate (PCC). PCC is indicated when acute restoration of oxygen-carrying capacity is required (i.e. to relieve symptomatic or predictably progressive anaemia) or to achieve marrow suppression in chronic ineffective erythropoiesis (e.g. thalassemia major, sickle cell anaemia). Nutritional anaemia rarely, if ever requires transfusion.
To calculate the desired transfusion volume, the following formula can be used: Packed red cells (mL) = weight (kg) × Hb rise required (g/L) × 0.4
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


