Type 1 diabetes mellitus
Diagnosis
Diagnosis is made by either:
- Random blood glucose >11 mmol/L, or
- Fasting blood glucose >7 mmol/L.
Note: There is no need for oral glucose tolerance testing.
Clinical features
- Typical symptoms are polyuria, polydipsia or weight loss. Glycosuria and ketonuria are often present.
- Children presenting with diabetes may range from being mildly unwell to severely unwell in diabetic ketoacidosis. Management varies according to presentation.
Differential diagnosis
Transient hyperglycaemia
Transient elevation of blood glucose and glycosuria (and possibly ketonuria) may occur in children with an intercurrent illness or with therapy such as glucocorticoids. The risk of later developing diabetes mellitus is about 3%, but it is approximately 30% if these findings are picked up in an otherwise well child. Check HbA1c and diabetes-related autoimmune markers (antibodies against insulin, glutamic acid decarboxylase (GAD) and islet cells) and discuss with a specialist.
Type 2 diabetes mellitus
This form of diabetes was rare in children. It is being seen increasingly in children who are overweight, those with a family history of type 2 diabetes mellitus and in some specific ethnic groups.
New presentation, mildly unwell
Assessment
Less than 3% dehydration, no acidosis and not vomiting.
Management
Initial treatment
- 0.25 units/kg of quick-acting insulin s.c. stat. Halve dose if <4 years old.
- If within 2 h of a meal give mealtime dose only (see below). Halve dose if <4 years old.
- Before breakfast and lunch give 0.25 units/kg of rapid-acting insulin. Before the evening meal give 0.25 units/kg rapid-acting insulin and 0.25 units/kg of intermediate-acting insulin. If this is the first insulin dose give 0.25 units/kg rapid-acting insulin only, then a further 0.25 units/kg rapid-acting insulin at midnight followed by a snack. Continue until normoglycaemia and negative ketonuria are achieved.
- Encourage fluid intake with sugar-free fluid and a normal diet according to appetite, but exclude foods with quick-acting sugars.
Ongoing treatment
- Once normoglycaemia is achieved and ketonuria disappears, change insulin to twice-daily mixtures of short and intermediate insulins. Several regimes and insulin types with varying profiles are available.
- The usual initial total dose is 1 unit/kg/day. This is given as:
– 2/3 in morning and 1/3 at night.
– 2/3 of each dose intermediate-acting and 1/3 as rapid-acting.
- Older adolescents often go on to a basal bolus regimen: 30–40% intermediate acting insulin given at 2200 h, remainder given as quick-acting insulin in three equal doses before meals.
- Other treatment modalities now include insulin pump therapy; see p. 307.
Hyperglycaemia, mildly unwell (known diabetic)
Usually advised to take 10% of total daily dose as rapid acting insulin every 2 h until normoglycaemic (in addition to normal insulin). Consult with a specialist if uncertain.
Diabetic ketoacidosis
This is the mode of presentation in >30% of newly diagnosed diabetes in childhood and adolescence. Diabetic ketoacidosis (DKA) may occur in any child or adolescent with established type 1 diabetes. Rapid onset is more likely in patients with poor underlying control or in patients on an insulin pump.
Definition
- Hyperglycaemia >14 mmol/L.
- Metabolic acidosis (pH <7.3 or bicarbonate <15 mmol/L).
- Hyperketonaemia or moderate to severe ketonuria.
Causes
- Delayed diagnosis of insulin-dependent diabetes mellitus (IDDM).
- Omission of insulin (especially in adolescents with recurrent DKA).
- Acute stress (infection, trauma, psychological).
- Poor management of intercurrent illness.
History
- Polyuria, polydipsia, loss of weight and lethargy. These symptoms are usually of 1–3 weeks’ duration in newly diagnosed patients. Symptoms are either absent or of shorter duration in patients with established diabetes.
- There may be a family history of diabetes or other autoimmune disease.
Examination
- Degree of dehydration (often overestimated):
– Mild/nil (<4%): no clinical signs.
– Moderate (4–7%): easily detectable dehydration (e.g. tachycardia, reduced skin turgor, poor capillary return).
– Severe (>7%) poor perfusion, rapid pulse, reduced blood pressure, i.e. shock.
- Altered level of consciousness.
- Body temperature – hypothermia is common.
- Presence of a precipitating cause (e.g. infection).
Investigations
- Blood glucose, urea and electrolytes.
- Arterial or capillary acid/base.
- Urine – ketones, culture.
- Check for precipitating cause e.g. infection (urine, FBE, blood cultures; consider chest radiograph).
- In all newly diagnosed patients: islet cell antibodies, insulin antibodies, GAD antibodies, total IgA, antiendomyseal IgA gliadin and transglutaminase antibodies and thyroid function tests.
- Calculate:
– Serum osmolality = 2Na+ + glucose + urea.
– Adjusted Na+ = plasma Na+ + 0.3(plasma glucose – 5.5).
Management
Initial fluid requirements
- If hypoperfusion is present, give normal saline at 10 mL/kg stat.
- Repeat until perfusion is re-established (warm, pink extremities with rapid capillary refill).
- Commence rehydration with normal saline (see Table 25.1).
- Keep nil by mouth (except ice to suck) until alert and stable.
- Insert a nasogastric tube if patient is comatose or has recurrent vomiting; leave on free drainage.
- Rehydration may be completed orally after the first 24–36 h if the patient is metabolically stable.
Table 25.1 Diabetic ketoacidosis fluid rates (mL/h) including deficit and maintenance fluid requirements, to be given evenly over 48 h
| Weight (kg) | Mild/Nil | Moderate |
| 5 | 24 | 27 |
| 7 | 33 | 38 |
| 8 | 38 | 43 |
| 10 | 48 | 54 |
| 12 | 53 | 60 |
| 14 | 58 | 67 |
| 16 | 64 | 74 |
| 18 | 70 | 80 |
| 20 | 75 | 87 |
| 22 | 78 | 91 |
| 24 | 80 | 95 |
| 26 | 83 | 100 |
| 28 | 86 | 104 |
| 30 | 89 | 108 |
| 32 | 92 | 112 |
| 34 | 95 | 116 |
| 36 | 98 | 120 |
| 38 | 101 | 125 |
| 40 | 104 | 129 |
| 42 | 107 | 133 |
| 44 | 110 | 137 |
| 46 | 113 | 141 |
| 48 | 116 | 146 |
| 50 | 119 | 150 |
| 52 | 122 | 154 |
| 54 | 124 | 158 |
| 56 | 127 | 162 |
| 58 | 130 | 167 |
| 60 | 133 | 171 |
| 62 | 136 | 175 |
| 64 | 139 | 179 |
| 66 | 142 | 183 |
| 68 | 145 | 187 |
| 70 | 148 | 191 |
Fluids once insulin is commenced
- Aim for maximum rate of fall of blood sugar of <5 mmol/L per hour. If the blood sugar falls more rapidly than this, very quickly, i.e. within the first few hours, change to normal saline (0.9%) with 5% dextrose.
- When the blood sugar reaches 12–15 mmol/L, use 0.45% NaCl with 5% dextrose. Aim to keep the blood sugar at 10–12 mmol/L.
- If the blood glucose falls below 10–12 mmol/L and the patient is still sick and acidotic, increase the dextrose in the infusate to 7.5–10%.
- Do not turn down insulin infusion.
Insulin
- Commence after treatment of shock.
- Add 50 units of clear/rapid-acting insulin (Actrapid or Humulin R) to 49.5 mL 0.9% NaCl (1 unit/mL solution).
- Ensure that the insulin is clearly labelled.
- Start at 0.1 unit/kg per hour in newly diagnosed children >2 years and those already on insulin who have glucose levels >15 mmol/L.
- Children who have had their usual insulin and whose blood sugars are <15 mmol/L and those <2 years of age should receive 0.05 units/kg per hour.
- Adjust the concentration of dextrose to keep blood glucose 10–12 mmol/L.
- Adequate insulin must be continued to clear acidosis (ketonaemia).
- Insulin infusion can be discontinued when the child is alert and metabolically stable (blood glucose <10–12 mmol/L, pH >7.30 and HCO3 >15 mmol/L). The best time to change to s.c. insulin is just before meal time.
- The insulin infusion should only be stopped 30 min after the first s.c. injection of insulin.
Potassium
- Add potassium chloride (KCl) to the i.v. fluid at the time of starting the insulin infusion.
- Start KCl at a concentration of 40 mmol/L if body weight <30 kg, or 60 mmol/L if ≥30 kg.
- Measure levels 2 h after starting therapy and 2–4 hourly thereafter.
- Specimens should be arterial or venous. Do not give K+ if the serum level is >5.5 mmol/L or if the patient is anuric.
Bicarbonate
- This is usually not necessary if shock has been adequately corrected. Continuing acidosis usually means insufficient resuscitation.
- In extremely sick children (with pH <7.0 ± HCO3 <5 mmol/L), small amounts may be given. Liaison with paediatric intensive care unit is advisable.
- The HCO3 dose (mmol) = 0.3 × body weight (kg) × base deficit. Infuse half over 30 mins with cardiac monitoring. Reassess acid–base status. Remember risk of hypokalaemia.
Monitoring
Clinical
- Strict fluid balance.
- Check all urine for ketones.
- Hourly observations: pulse, BP, respiratory rate and neurological observations.
- Hourly glucose (glucometer) while on insulin infusion.
- 4 hourly temperature.
Note: Any headache or altered behaviour may indicate impending cerebral oedema.
Biochemical
- 2–4 hourly laboratory blood glucose levels (with hourly bedside glucometer readings).
- Serum sodium (adjusted for hyperglycaemia), potassium, chloride.
- pH.
- Serum osmolality: should not fall >0.5 mmol/kg per hour.
Note: Beware of falling adjusted sodium levels as glucose declines – hyponatraemia may herald cerebral oedema. If the sodium level falls, consider decreasing the rate of fluid administration to replace over 72–96 h; see Hypernatraemia section below.
Other instructions
- Intensive care is required if age <2 years, coma, cardiovascular compromise or seizures.
- Patient should remain nil orally until alert and stable.
- Nurse the patient in a head-up position and in good light.
Complications of diabetic ketosis
Hypernatraemia
Measured serum sodium is depressed by the dilutional effect of the hyperglycaemia. If Na is >160 mmol/L, discuss with a specialist. Sodium should rise as the glucose falls during treatment. If this does not happen or if hyponatraemia develops, it usually indicates overzealous volume correction and insufficient electrolyte replacement. This may place the patient at risk of cerebral oedema.
Hypoglycaemia
If blood glucose <2.2 mmol/L give i.v. 25% dextrose 2 mL/kg over 3 min or 10% dextrose 5 mL/kg. Do not discontinue the insulin infusion. Continue with a 10% dextrose infusion until stable. Where hypoglycaemia is recurrent, increase concentration of dextrose in i.v. fluids (e.g. to 12.5% or 15%).
Note: 15% infusions require central access.
Hypokalaemia
Monitor frequently and adjust potassium concentration in the infusate. Children at particular risk of this complication are those who are very acidotic or have low potassium levels at presentation.
Cerebral oedema
This is an uncommon (0.5–3.0%) but extremely serious complication of diabetic ketoacidosis in children, usually occurring 6–12 h after commencement of therapy. This condition is often fatal. If the patient survives there may be profound neurological impairment.
Prevention
Slow correction of fluid and biochemical abnormalities. Optimally, the rate of fall of blood glucose and serum osmolality should not exceed 5 mmol/L per hour, but in children there is often a quicker initial fall in glucose. Patients should be nursed head up.
Risk factors
- Newly diagnosed diabetes, young age, poorly controlled diabetes.
- Excessive fluid rehydration, particularly with hypotonic fluids.
- Severe initial acidosis.
- Hyponatraemia or hypernatraemia and negative sodium trend during the therapy.
Note: With appropriate therapy the serum sodium should remain stable or rise slightly as blood glucose falls. If the adjusted serum sodium falls during resuscitation, this may be a sign of excess fluid administration and may be associated with the development of cerebral oedema. If this occurs, decrease the rate of fluid administration to replace over 72–96 h.
Signs
- Early: negative sodium trend, headache, behaviour change (sudden irritability, depression of conscious state) and incontinence.
- Late: bradycardia, elevated blood pressure and depressed respiration.
Treatment
Cerebral oedema is a medical emergency.
- Administer 20% mannitol i.v. as a bolus dose at 0.25–0.5 g/kg (1.25–2.5 mL/kg of 20%). This can be repeated if the response is inadequate.
- Nurse the patient in a head-up position, maintain the airway.
- Severely restrict fluids.
- Transfer to an intensive care unit for intubation, intermittent positive pressure ventilation and further management.
- Do not delay treatment for radiological confirmation – diagnosis is clinical.
Hypoglycaemia in children with diabetes Common causes
- Missed meal/snack.
- Vigorous exercise (can be during exercise or hours afterwards).
- Alcohol.
- Too much insulin.
Management
See Table 25.2
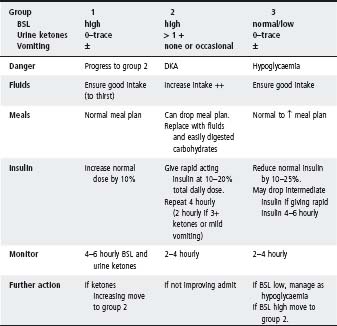
Sick day management during intercurrent illness in the child with diabetes
Principles
- Frequent testing of blood sugar and urine ketones (Table 25.3).
- The meal plan may temporarily be dropped – replace with fluids and easily digested carbohydrates.
- Ensure good fluid intake – alternate sugar and non-sugar-containing fluids depending on blood sugar levels (water is best if high).
- Insulin doses usually need to be increased; never omit insulin.
- Keep in touch with medical staff.
Management of children with diabetes undergoing surgery
The main aims are to prevent hypoglycaemia before, during and after surgery and to provide sufficient insulin to prevent the development of ketoacidosis. Factors that must be considered are:
- Time of surgery.
- Duration of surgery.
- Urgency of surgery.
Minor elective morning surgery
- Admit the child on the evening before surgery.
- Aim for the patient to be first on the operating list.
- Administer normal food and insulin until midnight on the night before surgery.
- At 0600 h check blood glucose. If blood glucose is <10 mmol/L give lemonade or sugar
containing clear fluid at 5–10 mL/kg (max = 200 mL) and inform the anaesthetist.
- Monitor blood glucose every 2 h and immediately before surgery. If blood glucose is <6 mmol/L insert i.v. line and give i.v. glucose.
- Give rapid-acting insulin equal to 1/10 of the total daily insulin dose (rapid- and intermediate-acting) at the usual time.
- An i.v. line with glucose will be inserted in the operating theatre (if not required preoperatively).
- Perform regular blood glucose every 2–4 h postoperatively, and adjust the i.v. glucose infusion as necessary. Give extra insulin 0.25 units/kg every 4–6 h to keep glucose between 5 and 10 mmol/L.
- When the patient can tolerate oral fluids stop the i.v. infusion and resume the normal insulin regimen.
Table 25.2 Management of hypoglycaemia in children with diabetes
| Awake | Sugar, e.g. 1 cup lemonade, orange or apple juice; jelly beans; honey (1 tbs); condensed milk in tube |
| Repeat in 5–10 min if no improvement, follow with ‘sustaining serve’, e.g. bread, milk | |
| Drowsy/uncooperative/unconscious/fitting (at home) | Glucagon* i.m. injection 1 mg (1 ampoule) if >25 kg or >8 years old (0.5 mg if <25 kg or <8 years) |
| Blood sugar rises in 5–10 min. Give sips of sugar-containing fluid when awake | |
| Uncooperative/unconscious/fitting (in hospital) | Glucose 2 mL/kg of 25% dextrose i.v. over 2 min, then infuse |
| 3–5 mg/kg per min until awake and able to eat/drink |
* Note: As glucagon can cause headache/vomiting, mini-glucagon rescue is preferred. It is used when vomiting is associated with hypoglycaemia or for persistently low blood glucose levels resistant to oral therapy. The doses vary with age: ≤2 years 0.02 mg; 2–15 years 0.01 mg/year of age; >15 years 0.15 mg.
Table 25.3 Sick day management
Minor elective afternoon surgery
- Continue normal food and insulin until midnight on the night before surgery.
- Provide a light breakfast at the usual time.
- Give rapid-acting insulin equal to 1/10 of the total daily insulin dose, half an hour before breakfast.
- Monitor blood glucose every 2 h (commence i.v. glucose if BSL is <6 mmol/L, otherwise i.v. glucose can be commenced in theatre).
- Give additional insulin at the same dose at 1200 h.
- Adopt same regimen as above postoperatively.
Minor surgery/short anaesthetic
I.v. glucose may not be necessary, provided that the oral intake can be resumed soon after surgery and that the pre-operative blood glucose concentration does not fall below 6 mmol/L. If in doubt, it is safer to follow the routines outlined above.
Emergency and major surgery
- Urgent clinical and biochemical assessment as for diabetic ketoacidosis.
- Rehydrate and start i.v. insulin as required.
- Maintain i.v. 0.45% saline with 5% dextrose and insulin infusion at 0.05–0.1 units/kg per hour pre- and postoperatively until the patient is able to resume oral feeding.
Continuous subcutaneous insulin infusion use in children and adolescents
Continuous subcutaneous insulin infusion (CSII), or insulin pump therapy, is gaining increasing use in the treatment of type 1 diabetes in children and adolescents. It relies on the continuous delivery of rapid-acting insulin into the subcutaneous tissues by an insulin pump, which is worn on the belt or in a bra, or carried in a pocket. The insulin pump is connected to a subcutaneous cannula by a dual-lumen flexible catheter. The subcutaneous cannula is re-sited and the pump reloaded with insulin every 3 days. Insulin pumps can be disconnected for 1– 2 h at a time for bathing, swimming or contact sports.
Indications
- CSII can be used at any age but is currently most common in adolescent patients.
- Although it offers benefits in terms of reduction of hypoglycaemia, improved glycaemic stability and improved quality of life, it is not suitable for all patients.
- At a minimum, patients must do 4–6 finger-prick blood glucose measures per day, be able to carbohydrate count accurately and be cognitively able to cope with the challenges of operating the insulin pump.
Calculating dose
Insulin is delivered in two ways:
- A continuous background delivery (basal delivery). Basal delivery is pre-programmed and automatic.
- An intermittent meal or correction-based insulin delivery (bolus delivery). Bolus delivery is manually undertaken by the patient at the time of meals or when blood glucose levels are to be corrected.
Total daily insulin dose (TDD) on CSII is derived from pre-existing insulin requirements or based on weight.
- 40–50% of the TDD is given as basal insulin and the remainder given as bolus insulin.
- Patients may have multiple basal rates at varying times of the day and according to the varying levels of activity.
- Bolus insulin doses are calculated using the ‘500’ rule (500 ÷ TDD = the number of grams of carbohydrate covered by 1 unit of bolus insulin). Correct calculation of the amount of bolus insulin requires that the patient is able to accurately ‘carb count’ their meals.
- The amount of insulin given to correct a high blood glucose level (correction factor) is calculated using the ‘100’ rule (100 ÷ TDD = the number of mmols drop in blood glucose that will result from a correction bolus of 1 unit of insulin).
- Most modern insulin pumps will automatically calculate bolus doses after they have been confi gured and a blood glucose level has been entered into the pump software.
Note: Infants and toddlers will usually require lower insulin bolus doses than are indicated by the ‘500’ and ‘100’ rules.
Complications
Diabetic ketoacidosis is the most concerning acute complication of CSII use (Table 25.4). These patients are at higher risk of DKA because they do not use any long- or intermediate-acting insulin.
If CSII therapy is discontinued it is critical that these patients have ongoing, regular and frequent review due to the risk of rapid onset of ketosis.
Stature must be assessed in the context of parental heights and pubertal status. Growth velocity must be compared with age-matched peers and assessed with regard to the child’s pubertal status. Consider: is a growth spurt occurring at an expected time for this child?
Stature
Measure the child and, wherever possible, both biological parents. Plot all three heights on appropriate height percentile charts, and compare the percentiles (see Appendix 1). For girls, adjust father’s height by subtracting 12.5 cm; for boys, adjust mother’s height by adding 12.5 cm. The child’s height percentile should approximate the mean of the parents’ percentiles.
Table 25.4 Complications of CSII use
| Complication | Causes | Management |
| Diabetic ketoacidosis | When insulin delivery is disrupted or when requirement increases. Ketosis ensues within 2–3 hours. | All patients receiving CSII therapy should also carry a back-up insulin pen containing rapid-acting insulin. |
| • Cannula dislodgement | • Hyperglycaemia – give correction bolus with pump | |
| • Tube kinks | • Recheck blood glucose in 1–2 h | |
| • Pump malfunction | • Glucose remains high and/or ketones in urine – disconnect pump and give | |
| • Intercurrent illness | ||
| • Infected insertion site | • CSII therapy can be recommenced once glycaemic stability and a lack of ketosis has been re-established | |
| Hypoglycaemia | • Excessive basal rate | • Temporarily cease pump therapy and give ‘hypo’ food |
| • Increased physical activity | • CSII should be recommenced as soon as normoglycaemia is re-established | |
| • Hypoglycaemia causing altered conscious state should be treated with glucagon as per the protocol (in addition to temporary cessation of CSII) |
Questions to consider:
- Is the child short in relation to other children the same age (i.e. below the third percentile)?
- Is the child unexpectedly short for the family?
Growth rate
Ask for any previous height measurements and plot them on the percentile chart. If no previous measurements are available, review at 3 month intervals; after 6 months, calculate the height velocity and check this against a growth velocity (GV) chart. Growth velocity can only be reliably calculated from measurements taken over 6–12 months. In most children, GV tends to fluctuate and only a consistently low GV will lead to a falling-off in height percentile. The criterion for further investigation in a short child is a GV below the 25th percentile.
Questions to consider:
- Is the child growing slowly?
- If the child’s growth really is slow, what is the reason?
- Is the child growing at the rate expected for pubertal status? A growth spurt should always accompany puberty.
Causes
Physiological
Constitutional delay in maturation
This is a common (and often familial) normal variant. Characteristically, growth slows at about 2 years of age, producing a fall in the height percentile. Thereafter, growth is parallel to the 3rd percentile, but the prepubertal decline in growth is exaggerated and the onset of the growth spurt is later than average. Bone age is delayed. The final height is likely to be in keeping with that of other family members.
Familial short stature
- Several adult family members are short. Skeletal proportions and GV are normal. Bone age is equivalent to the chronological age.
- Some children from short families also have constitutional delay in maturation. Parents who have suffered protein–calorie malnutrition as children may not have achieved their own genetic potential and may be on a lower percentile than their children.
Organic
Organic causes of short stature are classified in Table 25.5. Clues to the diagnosis may emerge from the history and the child’s general appearance. Some serious medical conditions (e.g. chronic renal failure, coeliac disease, inflammatory bowel disease, craniopharyngioma) may present with slow growth as the only abnormal sign. Important clues include:
- Dysmorphic features (e.g. Turner syndrome).
- Cutaneous changes (e.g. café au lait markings).
- Hand changes (e.g. short 4th/5th metacarpals, narrow deep-set nails).
- Fundal changes (e.g. optic atrophy).
Measure the skeletal proportions (arm span/height and upper/lower segment ratios).
- The lower segment should be >1/2 the height beyond the age of 8 years.
- The arm span should be within a few centimetres of height at all ages.
Investigations
Check the bone age initially. If the GV is <25th percentile for bone age then tests are indicated.
- Thyroid function tests – thyroid stimulating hormone (TSH) is the usual screening test, check free T4 (FT4) if central dysfunction is considered.
- Haemoglobin and erythrocyte sedimentation rate (ESR) (inflammatory bowel disease).
- Renal function and urine MC&S.
- Serum calcium, phosphate and alkaline phosphatase.
- Consider testing for coeliac disease.
- Chromosomes (all short girls; lack of dysmorphism does not exclude Turner syndrome).
- Skeletal survey (if disproportionate).
- Consider growth hormone (GH) studies as a second step (always done fasting):
Table 25.5 Organic causes of short stature
| Examples | Clues to diagnosis | |
| Intrauterine | Russell–Silver syndrome | Birth length <3rd centile for gestational age |
| Skeletal | Bone dysplasia (e.g. achondroplasia) | Skeletal disproportion (short limbs) |
| Spinal irradiation | ||
| Nutritional | Rickets | History of poor nutrition or limited sunlight/dark skin |
| Protein–calorie malnutrition (world no.1) | Low weight-for-height (if not chronic) | |
| Malabsorption (e.g. coeliac disease) | Abdominal distension | |
| Chronic illness (e.g. renal failure, Crohn’s disease) | Anaemia, high ESR | |
| Iatrogenic | Corticosteroid therapy | Cushingoid features |
| Chromosomal/genetic | Turner, Down, Prader–Willi, Noonan, Cornelia de Lange, Rubinstein–Taybi syndromes | Specific dysmorphic features |
| Inborn errors of metabolism: storage disorders (MPS, Gaucher) | ||
| Particular odour | ||
| Organic/amino acidopathies (e.g. MMA, MSUD) | Metabolic acidosis | |
| Endocrine | Hypothyroidism, Cushing disease, growth hormone deficiency, pubertal arrest, parathyroid disorders, AHO | Height centile < weight centile (i.e. short and plump) and associated examination fi ndings |
AHO, Albright hereditary osteodystrophy; MMA, methylmalonic aciduria; MPS, mucopolysaccharidosis; MSUD, maple syrup urine disease.
– Exercise.
– Glucagon stimulation (the current defi nitive test for GH deficiency).
Note: Basal GH is a useless test and does not defi ne GH deficiency.
Interventions
Growth hormone therapy
Recombinant human GH is government-controlled in Australia; it costs an average of $20 000–$30 000 per year per child. To qualify, children must meet certain criteria:
- Height is below the 1st percentile.
- GV is below the 25th percentile for bone age.
- Bone age is <13.5 years for girls, or <15.5 years for boys.
- Must be free of any condition known not to respond to GH (e.g. high-dose steroid therapy or thalassaemia) or that could be worsened by GH therapy (e.g. insulin-dependent diabetes mellitus (IDDM), Fanconi anaemia or active malignancy).
Children with GH deficiency or Turner syndrome respond to growth hormone with an increase in final height. Use of growth hormone for other conditions without biochemical GH deficiency will increase growth velocity in the short term but usually does not result in significant increase in final height.
Note: These criteria differ if a child has GH deficiency following intracranial pathology or treatment including cranial radiation. Specialist assessment is required.
The dose of growth hormone is 14–22 units/m2 per week divided into 6–7 doses/ week.
Causes
- Familial.
- Precocious puberty.
- Hyperthyroidism.
- Syndromes: Marfan, Klinefelter, triple X, homocystinuria and Sotos.
- Pituitary gigantism (juvenile acromegaly).
Assessment
Height must be considered in the context of midparental expectation and pubertal status, e.g. if puberty is 2–3 years earlier than average, the child may appear to be very tall for chronological age but have a perfectly normal final height expectation for the family.
Investigations
Consider:
- Thyroid function.
- Karyotype.
- Urine metabolic screen/antithrombin III/coagulation/lipids (homocystinuria).
- 3 h oral glucose tolerance test for GH/IGF1.
Management
Management of any underlying disorder, for example, precocious puberty. High-dose oestrogen is very seldom used in very tall girls, to hasten epiphyseal closure. Tall boys may be similarly treated with testosterone. Treatment is managed by a paediatric endocrinologist.
Hypothyroidism may be congenital or acquired.
Congenital hypothyroidism
Incidence is 1 : 3200 births.
Causes
- Absent thyroid 40–45%.
- Thyroid arrested in line of normal descent (lingual) 40–45%.
- Abnormal function (dyshormonogenesis) 10–15%.
- Thyroid hypolasia or hemithyroid (may be part of a genetic syndrome).
- Maternal iodine deficiency.
Clinical
- Unusually sleepy baby.
- Jaundice.
- Large anterior fontanelle, persistent posterior fontanelle.
- Coarse features.
- Dry skin.
- Periorbital oedema.
- Umbilical hernia.
- Harsh or hoarse cry.
- Slow feeding.
- Distal femoral epiphysis that is not ossified.
Investigations
- Most cases are detected by neonatal screening (high thyroid stimulating hormone (TSH)).
- Confirmation of the diagnosis on whole blood thyroid function tests (TFT) is essential.
- Technetium (Tch) scanning for position, function, size (presence of goitre, Tch uptake).
Management
Thyroxine therapy (8–12 mcg/kg per day) must be started as early as possible – before 2 weeks. Evidence suggests better outcome if treatment started at 10 days and T4 in upper range. Aim for:
- FT4 at the upper limit of normal range for age or just above.
- Normalisation of TSH.
Acquired hypothyroidism
Acquired hypothyroidism is called primary when the thyroid gland itself is abnormal (e.g. ectopic thyroid dysgenesis, autoimmune chronic lymphocytic thyroiditis and dyshormonogenesis) and secondary when the abnormality is a deficiency in pituitary TSH.
Clinical features
Hypothyroidism is often very difficult to detect clinically in children. Growth retardation may be the only sign, often with a relatively excess weight for height. The classical signs are usually absent when the cause is hypothalamic–pituitary.
• Growth retardation.
- Weight gain.
- Lethargy.
- Constipation.
- Cold intolerance.
- Goitre.
- Dry cool skin, dry hair.
- Prolonged ankle-jerk relaxation time.
Investigations
- TSH as screening test.
- FT4 for degree of deficit and for primary diagnosis when cause is central.
- Thyroid autoantibodies.
- Urinary iodine (early morning).
- Technetium thyroid scan.
- Thyroid ultrasound where indicated (for assessment of gland structure).
Note: Referral to a specialist is important for the management of hypothyroidism.
Hyperthyroidism is usually due to Graves’ disease in children and adolescents (different spectrum from that of adults). Six times as many girls are affected as boys, most commonly during puberty. A family history of thyroid disease (hyper- or hypothyroidism) is common. A family history should be sought for IDDM, vitiligo, pernicious anaemia, Addison disease or premature gonadal failure, as part of the spectrum of autoimmune polyglandular syndrome types I and II. Other causes to consider are:
- Toxic phase of Hashimoto thyroiditis (usually 4–6 weeks’ duration and usually not detected clinically).
- Thyroid adenoma (rare in childhood).
- Factitious (thyroxine consumption for weight loss).
Clinical features
- Goitre (nearly all), diffuse, with bruit.
- Weight loss, heat intolerance, tiredness.
- Warm sweaty hands, tremor, tachycardia.
- Irritability and restlessness.
- Proximal muscle weakness and wasting, accelerated ankle-jerk relaxation time.
- Lid lag; exophthalmos, peri-orbital oedema, extraocular muscle trapping causing diplopia on upward and lateral gaze.
- Accelerated growth velocity.
Investigations
- FT4, FT3. TSH should be suppressed to undetectable (<0.01 mU/L).
- TSH receptor antibodies.
- Bone age (usually advanced).
- Technetium thyroid scan – expect diffuse increased uptake.
- Thyroid ultrasound if adenoma suspected.
Management
Antithyroid drugs are used for long-term treatment in childhood and adolescence. The longterm remission rate in this age group is 40%. Management by a specialist is necessary.
Antithyroid drugs
- Carbimazole: 0.2 mg/kg (max 30–60 mg/day depending on age, size), oral 8–12 hourly, for 2 weeks. Then reduce dose to 0.1 mg/kg (max. 5 mg) oral 8–24 hourly for at least 18–24 months, until remission is achieved. Short courses of treatment result in low remission rates.
- Propylthiouracil (PTU): 5–7 mg/kg per day in three divided doses oral 8 hourly with similar reduction in dose after 2 weeks. Propylthiouracil prevents conversion of T4 to T3 and is the preferred treatment in severe toxicity.
Idiosyncratic reactions may occur to either drug with urticaria and/or neutropaenia. This can occur at any time during treatment but is more common with high doses early in treatment. There is approximately 40% crossover intolerance.
Surgery
Used for:
- Non-compliance.
- Allergy to drugs.
- Large goitre, increasing in size.
- Long-term patient choice.
Radioactive iodine
The use of radioactive iodine in children and young adolescents is controversial and is not advocated. WHO considers it safe after the age of 17 years. It is the treatment of choice for adults.
Thyroid storm
Thyroid storm is a rare complication of untreated primary hyperthyroidism or noncompliance with thyroid medication. It is characterised by tremor, anxiety, tachycardia, fever and confusion. It requires treatment (usually in ICU), with i.v. beta blockade, sedation, Lugol’s iodine and propylthiouracil.
Delayed puberty is defined as the absence of pubertal changes by 13–14 years for girls and by 15 years for boys. There is no absolute age for diagnosis; later than average and inappropriately late in a family being common reasons for referral (see Appendix 1 for pubertal stages charts/diagrams).
With normal or low serum gonadotrophins
- Constitutional delay (usually familial) is the most common cause. It is associated with slow growth and a delayed bone age in an otherwise healthy child.
- Chronic illness/poor nutrition (e.g. inflammatory bowel disease, anorexia nervosa, cystic fibrosis).
- Endocrine causes:
- – Hypopituitarism (gonadotrophin and possibly GH and other hormonal deficiencies). – Kallmann syndrome (isolated gonadotrophin deficiency with anosmia).
- – Hyperprolactinaemia (prolactinoma, secondary to medication (e.g. antipsychotics), functional (e.g. postcranial irradiation)).
With elevated serum gonadotrophins
This signifies primary gonadal failure, which may be due to:
- A genetic abnormality associated with gonadal dysgenesis (e.g. Turner, Klinefelter and Noonan syndromes).
- Anorchia.
- Gonadal destruction secondary to vascular damage, irradiation, infection, torsion or auto-immune disease.
Investigations
- Serum follicle-stimulating hormone (FSH), luteinising hormone (LH), testosterone or oestradiol; serum prolactin; other pituitary function tests (e.g. GH studies), as indicated by growth.
- Full blood examination, ESR.
- Urea, creatinine, serum proteins.
- Thyroid function test.
- Chromosomes.
- Bone age.
Management
Referral to a specialist is advised. Testosterone may be used in boys and oestradiol in girls; however, excess or too early use of sex hormones for pubertal management will result in rapid advancement of bone age, epiphyseal fusion and stunting of final height in both sexes. Growth hormone therapy may be offered to girls with Turner’s syndrome (see Short stature, pp. 309–312).
Precocious puberty is defined as the onset of pubertal changes under 8 years in girls and under 9.5 years in boys. For pubertal staging, see Appendix 1.
Cause
Gonadotrophin dependent (‘central’ or ‘true’ precocious puberty)
- True precocious puberty is 20 times more common in girls than boys.
- Girls are less likely to have an underlying pathological cause than boys.
- Girls with this disorder have accelerated growth, with development of both pubic hair and breasts, and the vaginal mucosa has a pale, shell-pink colour with increased mucus secretion due to the effects of oestrogen.
- Boys with true precocious puberty have enlargement of both testes, as well as accelerated linear and genital growth.
- The commonest pathological cause is hypothalamic hamartoma. Practically all intracranial pathologies (malformation, trauma, tumour, infection and haemorrhage) are associated with an increased prevalence of precocious puberty. After cranial irradiation, puberty occurs on average 2 years earlier than usual, in both boys and girls.
- Investigations are designed to demonstrate the premature activity in the hypothalamic– pituitary–gonadal axis and to exclude intracranial pathology.
Gonadotrophin independent (‘pseudo’ precocious puberty)
- Congenital adrenal hyperplasia.
- Adrenal, testicular or ovarian neoplasms.
- Tumours that secrete non-pituitary gonadotrophin such as chorionic gonadotrophin (hCG).
- McCune–Albright syndrome.
- Familial male precocious puberty.
Investigation of precocious puberty (both types)
- Serum FSH and LH.
- Gonadal steroid (testosterone or oestradiol).
- βHCG where indicated.
- Bone age.
- MRI of the head
– If increased FSH or LH is found.
– All boys with central precocious puberty must have an MRI scan.
– In girls with central precocious puberty MRI is less likely to yield an intracranial organic lesion. It is not commonly done if puberty occurs at >5 years, unless there is a specific clinical indication (e.g. headache, visual change).
- Pelvic ultrasound in girls for ovarian cyst or tumour.
- Testicular ultrasound if indicated.
Management
Refer to a specialist, who may use medroxyprogesterone acetate, cyproterone acetate or a luteinising hormone releasing hormone superagonist (LHRH agonist). Not all cases require treatment.
Conditions resembling precocious puberty
Premature thelarche
- Isolated breast development is common in girls <2 years of age (8–10%) and can be expected to regress spontaneously in most cases.
- Simple observation is usually sufficient but if the condition is associated with rapid growth velocity, consider true oestrogen excess of any cause and investigate (as above).
Premature adrenarche
- The isolated appearance of pubic hair (usually in a girl) under the age of 8 years may occur as a normal variant, but it may also signify non-classical congenital adrenal hyperplasia.
- Appropriate investigations are bone age, basal serum dehydroepiandrosterone sulfate (DHEA-S), androstenedione, testosterone and 17-hydroxyprogesterone (17-OHP). The measurement of 17-OHP at 30 and 60 min after intramuscular Synacthen (synthetic adrenocorticotrophic hormone (ACTH)) is recommended to diagnose non-classical congenital adrenal hyperplasia.
Note: Referral to a specialist is recommended.
- In true gynaecomastia there will be a palpable disc of breast tissue; this is to be distinguished from adiposity of the breast area.
- Breast development occurs transiently in many boys midway through puberty and is usually physiological.
- If associated with testicular volumes <6 mL, Klinefelter syndrome must be excluded by a chromosomal analysis (boys with Klinefelter syndrome are usually tall (>50th percentile), but this is not universal).
- Adrenal and gonadal tumours can cause gynaecomastia, but this is rare.
- Prolactinoma must be considered, particularly if gynaecomastia is associated with galactorrhoea.
- Many drugs, notably cimetidine, digoxin, spironolactone and i.m. testosterone can induce breast development, as can heavy use of marijuana.
- Prepubertal gynaecomastia also occurs, but in most cases no cause can be found.
Assessment
Most require no investigation, refer if concerned about diagnosis. Baseline FSH, LH, LFTs, prolactin, oestrogen can be helpful.
Management
90% of gynaecomastia disappears spontaneously within 2 years of onset. Refer boys with significant breast enlargement to a plastic or general surgeon for subareolar mastectomy. Tamoxifen is contraindicated in the young male as it inhibits oestrogen action and may delay epiphyseal fusion.
Table 25.6 Endocrine causes of obesity
| Cause | Clinical features | Screening investigation |
| Cushing disease | Growth retardation, hypertension, hirsutism, striae, typical facial changes, bruising | 24 h urinary free cortisol |
| Hypothyroidism | Growth retardation, tiredness, constipation, cold intolerance, dry skin | Thyroid function tests (TSH) |
| Growth hormone (GH) deficiency | Growth retardation | GH studies |
| Prader–Willi syndrome | Neonatal hypotonia, growth retardation, developmental delay, hyperphagia, hypogonadism, typical facial appearance, small hands and feet | Specific DNA FISH test |
See chapter 8, Obesity, and Table 25.6.
Nutritional obesity is associated with growth acceleration and advancement of bone age. Endocrine obesity is associated with growth retardation and a delay in bone age.
An underlying endocrine or genetic cause should be sought in:
- Any infant with ambiguous genitalia.
- Boys with perineal hypospadias.
- Boys with any combination of the following: micropenis, hypospadias, short stature, dysmorphic features or undescended testis.
- Girls with inguinal herniae containing gonads.
Note: Clitoral enlargement of any degree is abnormal.
Causes
In decreasing order of frequency:
- Gonadal dysgenesis.
- Congenital adrenal hyperplasia.
- Androgen insensitivity syndrome.
- Testosterone biosynthetic defects.
Investigations
- Electrolytes, urea and blood glucose.
- Serum 17-hydroxyprogesterone and 24 h urine steroid profile.
- Chromosomes.
- Pelvic ultrasound.
Management
- Refer urgently to an experienced paediatric endocrinologist and surgeon.
- Inform the parents about the problem and show them the genitalia; tell them that the infant appears otherwise healthy and that the true sex will be ascertained within a few days. Do not attempt to predict the child’s sex.
- Offer emotional support (refer to a social worker or an experienced mental health professional).
- Transfer the baby to a tertiary referral centre without delay.
- Call a meeting of all nursery staff and discuss policy about communication with the parents about the baby. Keep detailed notes about communication with the parents.
Primary adrenal insufficiency
This is rare in childhood and adolescence. It should be considered in the presence of vomiting, weight loss, pigmentation, chronic tiredness, low serum sodium and high serum potassium of unknown cause.
X-linked adrenoleukodystrophy
This is the commonest cause of primary adrenal insufficiency in school-age boys.
- Clinical hyperpigmentation of the skin (ACTH-mediated), tiredness, nausea, anorexia and weight loss.
- Adrenal features are usually but not always preceded by the development of a neurological disability (e.g. memory loss, sleep disturbance or ataxia).
- Test blood and skin fibroblasts for very-long-chain fatty acids.
- Dietary modification and bone marrow transplantation may be helpful in cases with normal MRI.
Autoimmune destruction (Addison disease)
- Autoimmune: This is usually part of the autoimmune polyglandular syndrome (in combination with either chronic mucocutaneous candidiasis, primary hypoparathyroidism, or both). Presenting in later childhood or adolescence, it may also be associated with thyrotoxicosis, diabetes mellitus, Hashimoto’s thyroiditis, coeliac disease, Graves disease, Sjögren syndrome, rheumatoid arthritis and less commonly with T or B cell deficiency.
- Infective: Worldwide, the commonest cause of Addison’s disease is TB, followed by HIV infection.
Congenital adrenal hyperplasia
- 21-Hydroxylase deficiency.
- Other rare types.
- Signs of androgen excess are usually obvious, with ambiguous genitalia and precocious sexual development.
Investigations
- Serum electrolytes (low sodium and high potassium).
- Simultaneous serum cortisol and plasma ACTH.
- Specific investigations if congenital adrenal hyperplasia (CAH) is suspected; 17-OH progesterone, urine steroid profile.
Management
- Hydrocortisone 12–20 mg/m2 BSA per day in divided doses.
- Fludrocortisone 0.05–0.2 mg daily, orally.
- Steroid cover for stress (see below).
‘Secondary’ adrenal insufficiency (due to ACTH deficiency)
Causes
Hypothalamic pituitary failure due to tumour, trauma, post surgery, cranial irradiation (where it may be subtle) or Langerhan’s histiocytosis.
Clinical
- Not usually associated with salt-wasting.
- No hyperpigmentation of the skin.
- Treat with hydrocortisone alone; fludrocortisone is unnecessary.
Steroid cover for stress (primary and secondary adrenal insuffi ciency)
- All patients with adrenal insuffi ciency of any cause are at risk for adrenal crisis during periods of severe stress.
- All need extra steroid cover.
- In cases of acute medical illness (e.g. gastroenteritis, influenza), any surgery requiring general anaesthetic and any major fracture:
– Hydrocortisone 0.2 mg–0.3 mg/kg (usually 25–100 mg) i.m./i.v. stat.
– Repeat every 4–6 h until recovery.
– Follow by triple the usual daily doses of hydrocortisone for 2 days, then double for 3 days.
Adrenocortical tumours
- This may manifest as Cushing syndrome, virilisation, hypertension, abdominal mass or pain.
- These tumours are very rare.
Adrenocortical hyperplasia
- This is usually secondary to a pituitary adenoma secreting ACTH (Cushing disease).
- The primary micronodular form (genetic cause) is rarely seen.
Cortisol excess is more diffi cult to detect clinically in children than in adults. It is characterised by poor growth velocity and excessive weight gain. The child usually looks obese but the clinical features of moon face, thin limbs and striae may be absent.
Investigation
- 24 h urinary free cortisol. Plasma cortisol is often abnormal in obesity and may give a spurious result.
- Overnight dexamethasone suppression (1 mg dexamethasone given at 2400 h and a plasma cortisol at 0800 h the following day) will differentiate Cushing syndrome from obesity.
Further investigation for origin and type of cortisol excess is by a specialist. Treatment is surgical.
Adrenal medullary tumours
- Neuroblastoma usually occurs in very young children, but may present in adolescence.
- Phaeochromocytoma in older children (leading to hypertension).
Note: Phaeochromocytoma may be associated with various genetic conditions – MEN, neurofi bromatosis, von Hippel Lindau, SDH mutations.
Disorders of calcium metabolism
Hypocalcaemia
For causes, see Table 25.7.
Clinical features
- Rachitic changes in long bones (swollen wrists, etc.), rachitic rosary.
- Tetany (may be demonstrated using sphygmomanometer cuff above systolic pressure for up to 2 min).
- Laryngeal stridor.
- Fitting.
- Weakness, tiredness, irritability.
- Even extreme hypocalcaemia may be asymptomatic in an infant.
Investigations
- 25-OH vitamin D.
- Renal function, lipase, albumin.
- Magnesium, phosphate.
- Alkaline phosphatase.
- Parathyroid hormone (PTH).
- Total and ionised calcium.
- 1,25-diOH-vitamin D if hypophosphataemic rickets is suspected.
- Radiographs of wrist, knee (metaphyseal splaying).
- Malabsorption studies.
- ECG (prolonged QT interval).
Treatment
Emergency
- I.v. calcium chloride 10% (infusion 1 mmol/kg per 24 h in 5% dextrose), monitor calcium levels 6 hourly.
- Occasionally i.v. calcium chloride 10%, 0.2 mL/kg stat may be required for severe tetany.
Table 25.7 Causes of hypocalcaemia
| Neonatal presentation | Infant/childhood presentation |
| Prematurity/IUGR/ birth asphyxia | Vitamin D defi ciency. Groups at risk: |
| Hypoparathyroidism ± | • Families where covering clothing is worn at all times, especially breast-fed infants in such families |
| Di George syndrome | • dark skin colour |
| Phosphate load (high phosphate milk) | • Indoor lifestyle |
| Low magnesium | • Anticonvulsants (alter vitamin D metabolism/calcium absorption) |
| Chronic immobilisation | |
| • Malabsorption, liver disease | |
| Hypoparathyroidism | |
| • Association with autoimmune polyglandular syndrome. Look for mucocutaneous candidiasis and/or Addison disease in a young child | |
| Pseudohypoparathyroidism | |
| • Albright hereditary osteodystrophy | |
| Chronic renal failure | |
| Pancreatitis | |
| Organic acidaemia | |
| Critical illness | |
| 1-α-hydroxylase deficiency (rare) | |
| Vitamin D resistant rickets |
- Correct magnesium if low.
- ECG monitor.
- 25-OH-vitamin D if nutritional rickets is suspected: 4000 IU/day for 2 weeks then maintenance (see below).
- 1,25-diOH-vitamin D if parathyroid disorders suspected: 0.01–0.02 mcg/kg per day starting dose may need to be increased.
- Treatment of underlying condition.
Note: In the first days to weeks after treatment is begun for rickets, bones are ‘hungry’ – large doses of calcium supplement may be required to maintain normocalcaemia and prevent carpopedal spasm once vitamin D is started.
Maintenance
- Adequate calcium intake, preferably as dairy products, 600–1500 mg/day depending on age.
- 500 IU/day 25-OH-vitamin D for months–years depending on cause (for infant rickets, usually treat to age 4).
- Stoss therapy is alternative method, using 100 000–150 000 units 25-OH-vitamin D every 3–6 months as required. Monitoring is required.
- 1,25-diOH-vitamin D (Rocaltrol) for vitamin D resistant rickets, hypoparathyroidism, patients on anticonvulsants.
- 1,25-diOH-vitamin D and phosphate for hypophosphataemic rickets (high dose).
Hypercalcaemia
For causes, see Table 25.8.
Clinical features
- Polyuria, polydipsia.
- Vomiting, dehydration.
- Failure to thrive.
- Abdominal pain (constipation, renal stones, pancreatitis).
- Confusion, apathy (if severe).
Investigation
- Total and ionised calcium, phosphate.
- Magnesium, albumin.
- Alkaline phosphatase.
- 25-OH-vitamin D ± 1,25-diOH-vitamin D.
- Thyroid function.
- Chest radiograph ± skeletal survey.
- Parathyroid imaging.
- Renal ultrasound (nephrocalcinosis).
- ECG (short QT interval).
Table 25.8 Causes of hypercalcaemia
| Neonatal | Infant/childhood |
| Hyperparathyroidism (rare) Iatrogenic | Primary hyperparathyroidism |
| Subcutaneous fat necrosis | Familial hypocalciuric hypercalcaemia |
| Vitamin D 1,25-diOH-D excess (nutritional, inflammatory disease e.g. sarcoidosis, leukaemia) | Familial hypocalciuric hypercalcaemia – severe form |
| Hypophosphatasia | Neoplasia (lytic bone lesions or humoral hypercalcaemia PTHrP) |
| Bartter syndrome | Immobilisation e.g. burns (severe), quadriplegia – can be very severe and cause renal calculi, pancreatitis |
| William syndrome (with elfi n face, supravalvular aortic stenosis) | Drugs (lithium, thiazides) |
| Endocrine disorders: Hyperthyroidism (mild, usually asymptomatic), phaeochromocytoma, adrenal insufficiency |
Management of severe hypercalcaemia
- Rehydration with 0.9% saline/5% dextrose. For infants <2 years use 0.45% saline/5% dextrose.
- Diuretics (thiazide) are used in two situations:
– Acute hypercalcaemia: frusemide is given to reduce fluid overload while the patient is aggressively rehydrated.
– Chronic hypercalcaemia with hypercalciuria (or normocalcaemia with hypercalciuria): thiazides are given to reduce oedema and prevent nephrocalcinosis (by decreasing urinary calcium excretion).
- Bisphosphonates, particularly for increased bone resorption (e.g. immobilisation).
- Steroids for vitamin D excess (prednisolone 2 mg/kg per day, reducing).
- Low-calcium diet.
- Surgery if indicated, treatment of underlying condition.
USEFUL RESOURCES
- www.apeg.org.au – Australian Paediatric Endocrine Group includes downloadable booklets for patients.
- www.rch.org.au/chas/pubs – Complete Androgen Insensitivity Syndrome by Garry Warne.
- www.magicfoundation.org – Major Aspects of Growth in Children, a USA-based organisation supporting families and children with conditions affecting growth.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


